
Development of a Target Enrichment Probe Set for Conifer (REMcon)
 , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Probe Design
2.2. Taxon Selection
2.3. DNA Extraction, Library Preparation, Hybrid Capture and Sequencing
2.4. Bioinformatics Analyses
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ran, J.-H.; Gao, H.; Wang, X.-Q. Fast evolution of the retroprocessed mitochondrial rps3 gene in Conifer II and further evidence for the phylogeny of gymnosperms. Mol. Phylogenetics Evol. 2010, 54, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ferguson, D.K.; Liu, B.; Mao, K.S.; Gao, L.M.; Zhang, S.Z.; Wan, T.; Rushforth, K.; Zhang, Z.X. Recent advances on phylogenomics of gymnosperms and an updated classification. Plant Divers. 2022, 44, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Hill, R.S.; Liu, J.; Biffin, E. Diversity, Distribution, Systematics and Conservation Status of Podocarpaceae. Plants 2023, 12, 1171. [Google Scholar] [CrossRef] [PubMed]
- Armenise, L.; Simeone, M.C.; Piredda, R.; Schirone, B. Validation of DNA barcoding as an efficient tool for taxon identification and detection of species diversity in Italian conifers. Eur. J. For. Res. 2012, 131, 1337–1353. [Google Scholar] [CrossRef]
- Leslie, A.B.; Beaulieu, J.; Holman, G.; Campbell, C.S.; Mei, W.; Raubeson, L.R.; Mathews, S. An overview of extant conifer evolution from the perspective of the fossil record. Am. J. Bot. 2018, 105, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Hill, R.S.; Dörken, V.M.; Biffin, E. Detailed seed cone morpho-anatomy of the Prumnopityoid clade: An insight into the origin and evolution of Podocarpaceae seed cones. Ann. Bot. 2022, 130, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Hill, R.S.; Dörken, V.M.; Biffin, E. Detailed Seed Cone Morpho-Anatomy Provides New Insights into Seed Cone Origin and Evolution of Podocarpaceae; Podocarpoid and Dacrydioid Clades. Plants 2023, 12, 3903. [Google Scholar] [CrossRef]
- Kelch, D.G. Phylogeny of Podocarpaceae: Comparison of evidence from morphology and 18S rDNA. Am. J. Bot. 1998, 85, 986–996. [Google Scholar] [CrossRef]
- Conran, J.G.; Wood, G.M.; Martin, P.G.; Dowd, J.M.; Quinn, C.J.; Gadek, P.A.; Price, R.A. Generic relationships within and between the gymnosperm families Podocarpaceae and Phyllocladaceae based on an analysis of the chloroplast gene rbcL. Aust. J. Bot. 2000, 48, 715–724. [Google Scholar] [CrossRef]
- Sinclair, W.; Mill, R.; Gardner, M.; Woltz, P.; Jaffré, T.; Preston, J.; Hollingsworth, M.; Ponge, A.; Möller, M. Evolutionary relationships of the New Caledonian heterotrophic conifer, Parasitaxus usta (Podocarpaceae), inferred from chloroplast trn LF intron/spacer and nuclear rDNA ITS2 sequences. Plant Syst. Evol. 2002, 233, 79–104. [Google Scholar] [CrossRef]
- Knopf, P.; Schulz, C.; Little, D.P.; Stützel, T.; Stevenson, D.W. Relationships within Podocarpaceae based on DNA sequence, anatomical, morphological, and biogeographical data. Cladistics 2012, 28, 271–299. [Google Scholar] [CrossRef] [PubMed]
- Little, D.P.; Knopf, P.; Schulz, C. DNA barcode identification of Podocarpaceae—The second largest conifer family. PLoS ONE 2013, 8, e81008. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, M.R.; Neale, D.B. Evolution of genome size in conifers. Silvae Genet. 2005, 54, 126–137. [Google Scholar] [CrossRef]
- Zonneveld, B.J.M. Conifer genome sizes of 172 species, covering 64 of 67 genera, range from 8 to 72 picogram. Nord. J. Bot. 2012, 30, 490–502. [Google Scholar] [CrossRef]
- Weitemier, K.; Straub, S.C.; Cronn, R.C.; Fishbein, M.; Schmickl, R.; McDonnell, A.; Liston, A. Hyb-Seq: Combining target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci. 2014, 2, 1400042. [Google Scholar] [CrossRef] [PubMed]
- Vatanparast, M.; Powell, A.; Doyle, J.J.; Egan, A.N. Targeting legume loci: A comparison of three methods for target enrichment bait design in Leguminosae phylogenomics. Appl. Plant Sci. 2018, 6, e1036. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.G.; Pokorny, L.; Dodsworth, S.; Botigue, L.R.; Cowan, R.S.; Devault, A.; Eiserhardt, W.L.; Epitawalage, N.; Forest, F.; Kim, J.T.; et al. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst. Biol. 2019, 68, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Breinholt, J.W.; Carey, S.B.; Tiley, G.P.; Davis, E.C.; Endara, L.; McDaniel, S.F.; Neves, L.G.; Sessa, E.B.; von Konrat, M.; Chantanaorrapint, S.; et al. A target enrichment probe set for resolving the flagellate land plant tree of life. Appl. Plant Sci. 2021, 9, e11406. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Schneider, J.V.; Zizka, G.; Maurin, O.; Baker, W.; Forest, F.; Brewer, G.E.; Savolainen, V.; Darbyshire, I.; Larridon, I. Joining forces in Ochnaceae phylogenomics: A tale of two targeted sequencing probe kits. Am. J. Bot. 2021, 108, 1201–1216. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.; Dodsworth, S.; Forest, F.; Graham, S.; Johnson, M.; McDonnell, A.; Pokorny, L.; Tate, J.A.; Wicke, S.; Wickett, N. Exploring Angiosperms353: An open, community toolkit for collaborative phylogenomic research on flowering plants. Am. J. Bot. 2021, 108, 1059–1065. [Google Scholar] [CrossRef]
- Zuntini, A.R.; Carruthers, T.; Maurin, O.; Bailey, P.C.; Leempoel, K.; Brewer, G.E.; Epitawalage, N.; Françoso, E.; Gallego-Paramo, B.; Baker, W.J.; et al. Phylogenomics and the rise of the angiosperms. Nature, 2024; Online ahead of print. [Google Scholar] [CrossRef]
- Léveillé-Bourret, É.; Starr, J.R.; Ford, B.A.; Moriarty Lemmon, E.; Lemmon, A.R. Resolving rapid radiations within angiosperm families using anchored phylogenomics. Syst. Biol. 2018, 67, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Montes, J.R.; Peláez, P.; Willyard, A.; Moreno-Letelier, A.; Piñero, D.; Gernandt, D.S. Phylogenetics of Pinus subsection Cembroides Engelm. (Pinaceae) inferred from low-copy nuclear gene sequences. Syst. Bot. 2019, 44, 501–518. [Google Scholar] [CrossRef]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Jansson, S.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Shalev, T.J.; El-Dien, O.G.; Yuen, M.M.; Shengqiang, S.; Jackman, S.D.; Warren, R.L.; Coombe, L.; van der Merwe, L.; Stewart, A.; Bohlmann, J.; et al. The western redcedar genome reveals low genetic diversity in a self-compatible conifer. Genome Res. 2022, 32, 1952–1964. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Hill, R.S. Reproductive and leaf morpho-anatomy of the Australian alpine podocarp and comparison with the Australis subclade. Bot. Lett. 2022, 169, 237–249. [Google Scholar] [CrossRef]
- Khan, R.; Hill, R.S. Morpho-anatomical affinities and evolutionary relationships of three paleoendemic podocarp genera based on seed cone traits. Ann. Bot. 2021, 128, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Wall, P.K.; Edger, P.P.; Landherr, L.L.; Ma, H.; Pires, P.K.; Leebens-Mack, J.; dePamphilis, C.W. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010, 10, 1–18. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17, 282–283. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Hancock-Hanser, B.L.; Frey, A.; Leslie, M.S.; Dutton, P.H.; Archer, F.I.; Morin, P.A. Targeted multiplex next-generation sequencing: Advances in techniques of mitochondrial and nuclear DNA sequencing for population genomics. Mol. Ecol. Resour. 2013, 13, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Hugall, A.F.; O’Hara, T.D.; Hunjan, S.; Nilsen, R.; Moussalli, A. An exon-capture system for the entire class Ophiuroidea. Mol. Biol. Evol. 2015, 33, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Waycott, M.; van Dijk, J.K.; Biffin, E. A hybrid capture RNA bait set for resolving genetic and evolutionary relationships in angiosperms from deep phylogeny to intraspecific lineage hybridization. bioRxiv 2022. [Google Scholar] [CrossRef]
- Andermann, T.; Cano, Á.; Zizka, A.; Bacon, C.; Antonelli, A. SECAPR—A bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ 2018, 6, e5175. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Yang, Y.; Smith, S.A. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: Improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 2014, 31, 3081–3092. [Google Scholar] [CrossRef]
- Jackson, C.; McLay, T.; Schmidt-Lebuhn, A.N. hybpiper-nf and paragone-nf: Containerization and additional options for target capture assembly and paralog resolution. Appl. Plant Sci. 2023, 11, e11532. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- McLay, T.G.; Birch, J.L.; Gunn, B.F.; Ning, W.; Tate, J.A.; Nauheimer, L.; Joyce, E.M.; Simpson, L.; Schmidt-Lebuhn, A.N.; Baker, W.J.; et al. New targets acquired: Improving locus recovery from the Angiosperms353 probe set. Appl. Plant Sci. 2021, 9, e11420. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Baniaga, A.E.; Sessa, E.B.; Scascitelli, M.; Graham, S.W.; Rieseberg, L.H.; Barker, M.S. Early genome duplications in conifers and other seed plants. Sci. Adv. 2015, 1, e1501084. [Google Scholar] [CrossRef] [PubMed]
- Stull, G.W.; Qu, X.J.; Parins-Fukuchi, C.; Yang, Y.Y.; Yang, J.B.; Yang, Z.Y.; Hu, Y.; Ma, H.; Soltis, P.S.; Soltis, D.E.; et al. Gene duplications and phylogenomic conflict underlie major pulses of phenotypic evolution in gymnosperms. Nat. Plants 2021, 7, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.G. Nuclear DNA amounts in gymnosperms. Ann. Bot. 1998, 82 (Suppl. 1), 3–15. [Google Scholar] [CrossRef]
- Kinlaw, C.S.; Neale, D.B. Complex gene families in pine genomes. Trends Plant Sci. 1997, 2, 356–359. [Google Scholar] [CrossRef]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving difficult phylogenetic questions: Why more sequences are not enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef]
- Whitfield, J.B.; Lockhart, P.J. Deciphering ancient rapid radiations. Trends Ecol. Evol. 2007, 22, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Mongiardino Koch, N. Phylogenomic subsampling and the search for phylogenetically reliable loci. Mol. Biol. Evol. 2021, 38, 4025–4038. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, W.T.; Liu, X.Q.; Wang, X.Q. New insights into the phylogeny and evolution of Podocarpaceae inferred from transcriptomic data. Mol. Phylogenetics Evol. 2022, 166, 107341. [Google Scholar] [CrossRef] [PubMed]
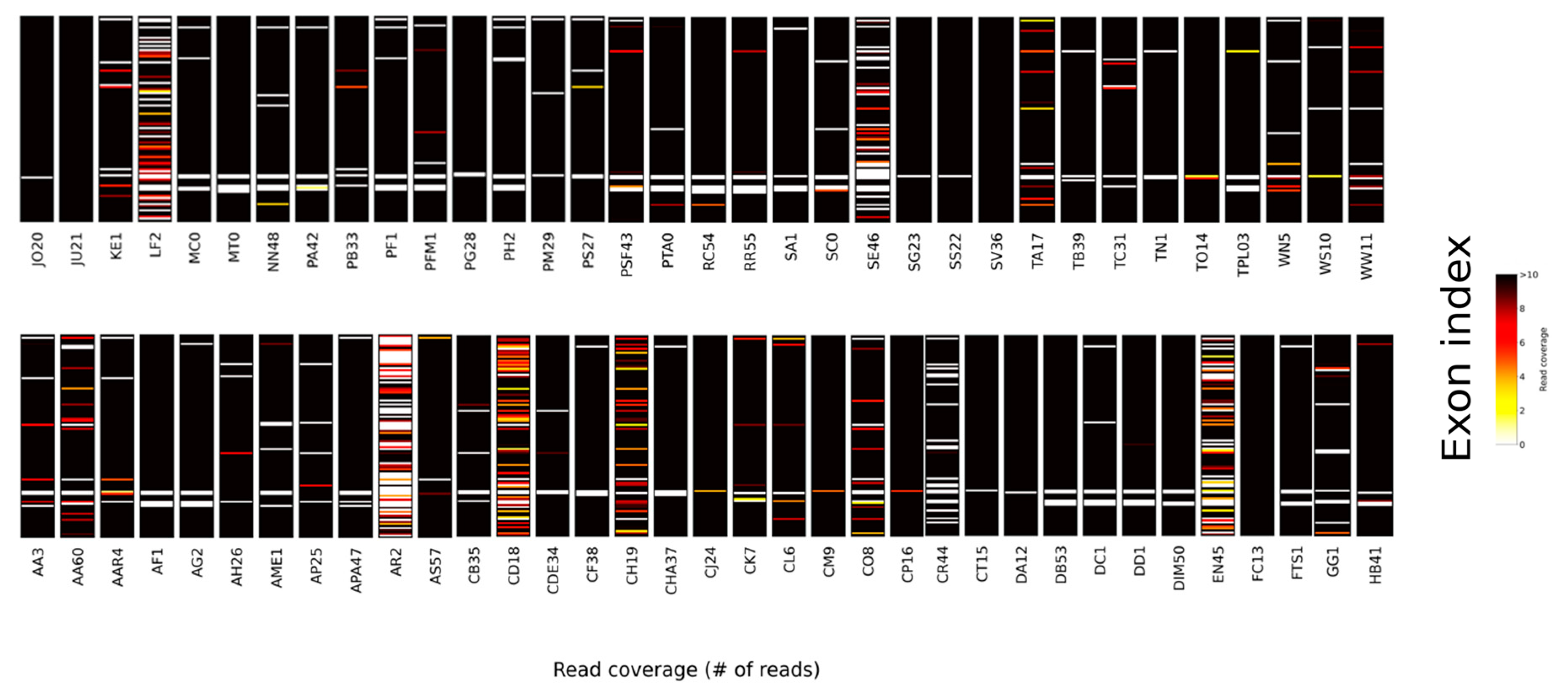
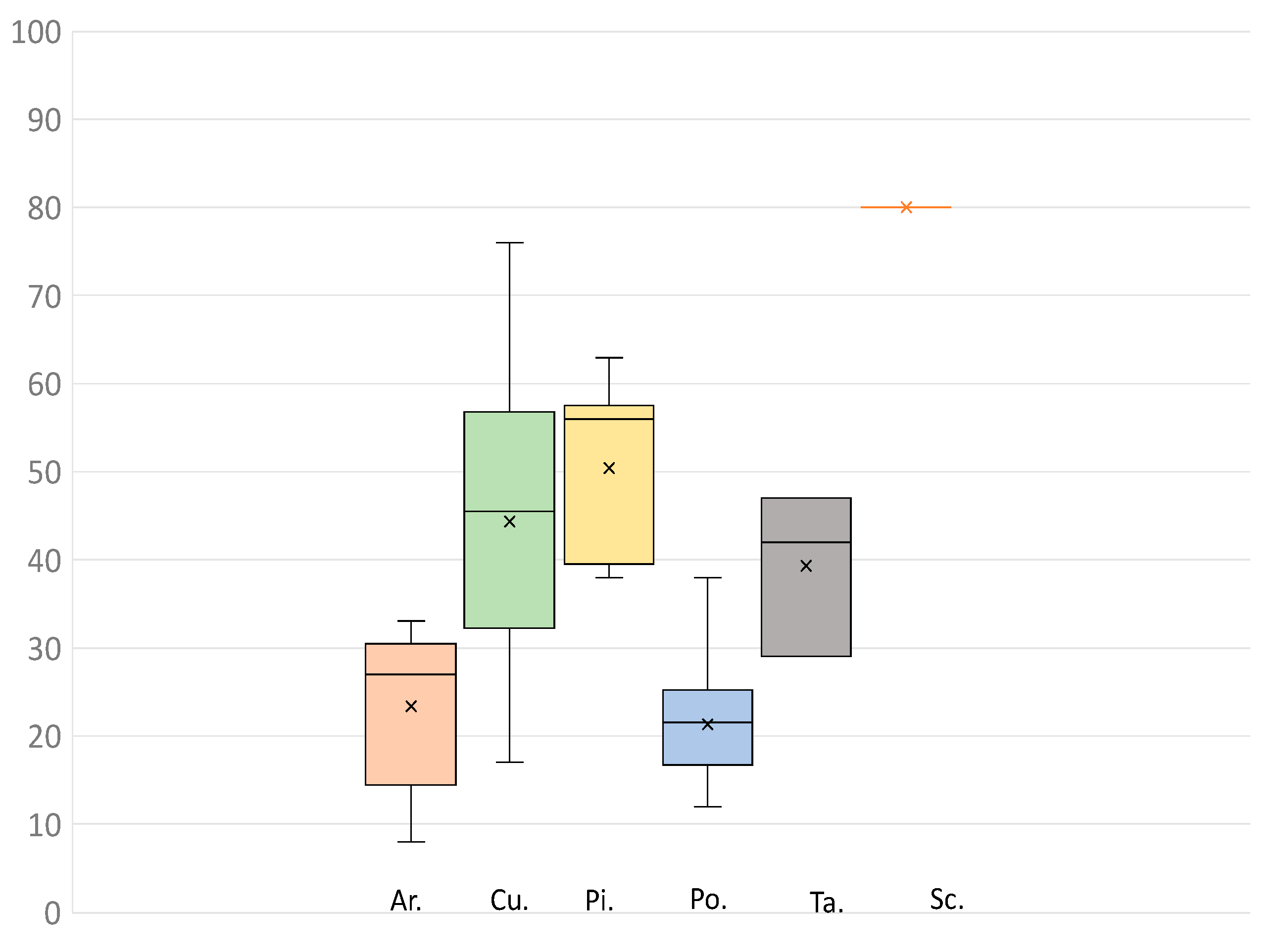
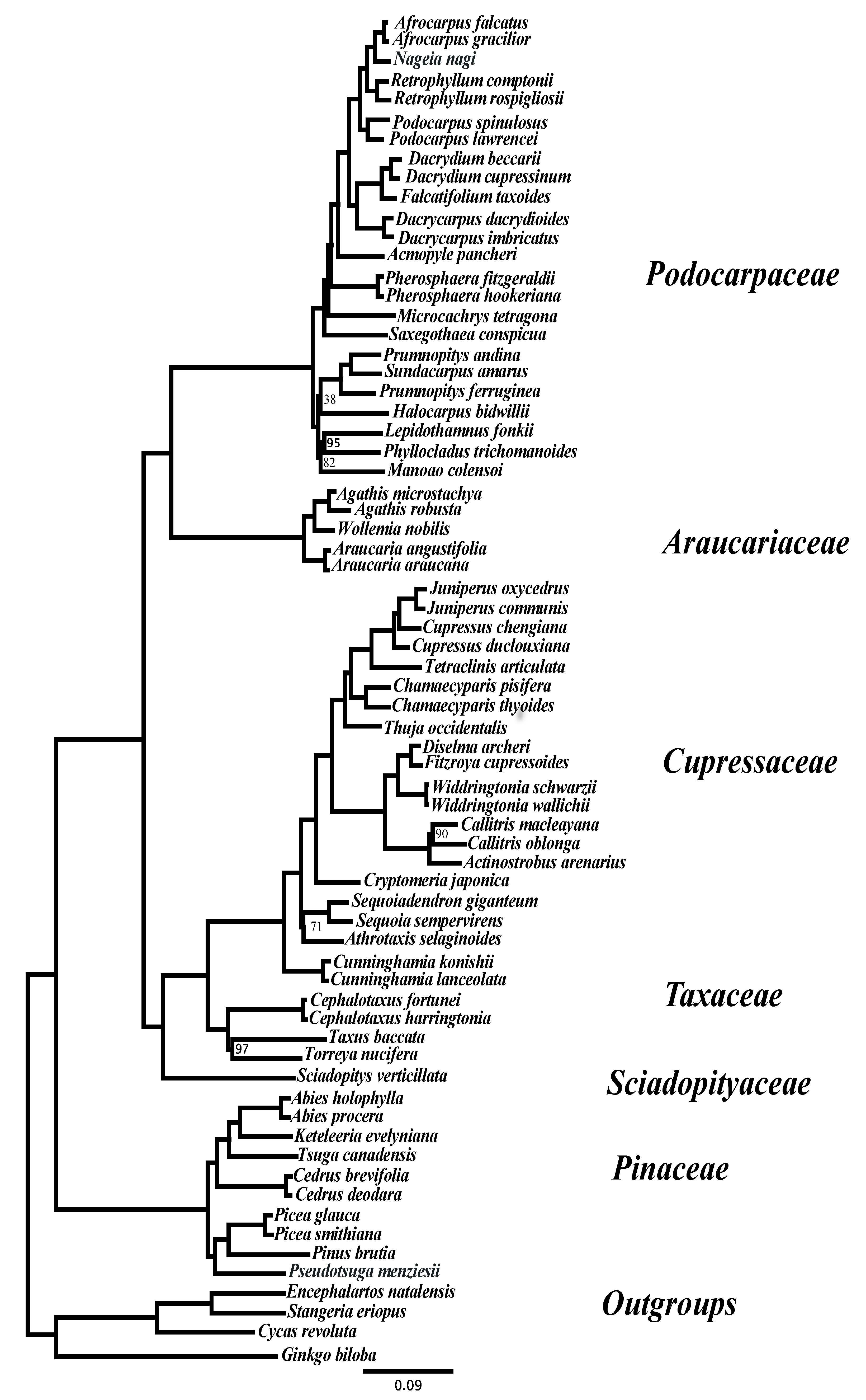

{kind=link}
{kind=link}
{kind=link}
| S# | Picea abies Gene Name | Arabidopsis thaliana Putative Homolog | Length of the Probe Sequences |
|---|---|---|---|
| 1 | MA_10437158 | AT5G06430 | 195 |
| 2 | MA_10437143 | AT1G12370 | 480 |
| 3 | MA_10437077 | AT5G02250 | 621 |
| 4 | MA_10437070 | AT5G10920 | 720 |
| 5 | MA_10436603 | AT1G03750 | 945 |
| 6 | MA_10436489 | AT4G37510 | 878 |
| 7 | MA_10435966 | AT4G38890 | 822 |
| 8 | MA_10435879 | AT2G33630 | 613 |
| 9 | MA_10435851 | AT5G04520 | 510 |
| 10 | MA_10435433 | AT2G44760 | 426 |
| 11 | MA_10435005 | AT2G40570 | 787 |
| 12 | MA_10434812 | AT1G36310 | 1088 |
| 13 | MA_10434753 | AT1G49380 | 539 |
| 14 | MA_10433768 | AT2G31955 | 942 |
| 15 | MA_10433107 | AT5G64150 | 825 |
| 16 | MA_10432498 | AT1G74640 | 453 |
| 17 | MA_10431375 | AT2G24830 | 321 |
| 18 | MA_10430781 | AT4G35910 | 432 |
| 19 | MA_10429426 | AT1G30070 | 240 |
| 20 | MA_10428930 | AT1G15390 | 256 |
| 21 | MA_10428614 | AT2G34640 | 259 |
| 22 | MA_10428345 | AT1G57770.1 | 315 |
| 23 | MA_10428134 | AT2G04560 | 273 |
| 24 | MA_10427767 | AT1G21370 | 291 |
| 25 | MA_10427729 | AT5G67530 | 1224 |
| 26 | MA_10427590 | AT1G17160 | 480 |
| 27 | MA_10427203 | AT2G36740 | 543 |
| 28 | MA_10426631 | AT4G36390 | 1533 |
| 29 | MA_10426581 | AT2G33450 | 231 |
| 30 | MA_10426376 | AT2G38270 | 504 |
| 31 | MA_9578808 | AT4G18372 | 387 |
| 32 | MA_9514062 | AT5G20220 | 315 |
| 33 | MA_9503281 | AT1G48175 | 257 |
| 34 | MA_8815984 | AT2G346401 | 693 |
| 35 | MA_8715484 | AT4G38020 | 501 |
| 36 | MA_8687206 | AT4G26980 | 408 |
| 37 | MA_8286794 | AT3G17170 | 342 |
| 38 | MA_8140147 | AT2G28605 | 480 |
| 39 | MA_7890741 | AT2G44660 | 783 |
| 40 | MA_5587080 | AT4G20060 | 447 |
| 41 | MA_957334 | AT1G05055 | 462 |
| 42 | MA_945784 | AT5G06410 | 380 |
| 43 | MA_939779 | AT4G27390 | 468 |
| 44 | MA_938037 | AT5G49570 | 580 |
| 45 | MA_894439_ | AT2G30100 | 1306 |
| 46 | MA_824260 | AT4G28020 | 441 |
| 47 | MA_762004 | AT1G28560 | 675 |
| 48 | MA_759516 | AT5G08720 | 461 |
| 49 | MA_749379 | AT4G11980 | 201 |
| 50 | MA_587488 | AT4G01040 | 377 |
| 51 | MA_546546 | AT4G17760 | 252 |
| 52 | MA_537299 | AT5G54840 | 264 |
| 53 | MA_458270 | AT5G06830 | 690 |
| 54 | MA_388031 | AT2G20330 | 486 |
| 55 | MA_341112 | AT5G11980 | 276 |
| 56 | MA_332596 | AT2G34460 | 333 |
| 57 | MA_314789 | AT1G56345.1 | 603 |
| 58 | MA_261436 | AT4G33030 | 1290 |
| 59 | MA_253636 | AT3G51050 | 768 |
| 60 | MA_225872 | AT5G14260 | 456 |
| 61 | MA_224167 | AT2G20790 | 900 |
| 62 | MA_199851 | AT3G01660 | 350 |
| 63 | MA_196209 | AT4G36530 | 273 |
| 64 | MA_187402 | AT4G31460 | 471 |
| 65 | MA_173127 | AT4G28740 | 548 |
| 66 | MA_159115 | AT2G27600 | 1191 |
| 67 | MA_159115 | AT4G27600 | 1056 |
| 68 | MA_127668 | AT3G15290 | 465 |
| 69 | MA_123340 | AT2G19870 | 1137 |
| 70 | MA_121485 | AT1G02410 | 749 |
| 71 | MA_121026 | AT1G08460 | 570 |
| 72 | MA_106933 | AT2G266801 | 636 |
| 73 | MA_104872 | AT3G26580 | 507 |
| 74 | MA_99242 | AT4G29070 | 412 |
| 75 | MA_98424 | AT1G07130 | 558 |
| 76 | MA_95157 | AT5G09820 | 292 |
| 77 | MA_83545 | AT5G65860 | 514 |
| 78 | MA_78599 | AT2G40760 | 252 |
| 79 | MA_73742 | AT2G21840 | 939 |
| 80 | MA_73742 | AT1G21840 | 939 |
| 81 | MA_67861 | AT2G26680 | 369 |
| 82 | MA_66902 | AT2G36145 | 234 |
| 83 | MA_66902 | AT2G34145 | 234 |
| 84 | MA_63465 | AT3G24315 | 290 |
| 85 | MA_61548 | AT1G65030 | 681 |
| 86 | MA_55048 | AT5G19130 | 858 |
| 87 | MA_43083 | AT5G48330 | 717 |
| 88 | MA_41847 | AT3G03790 | 303 |
| 89 | MA_35149 | AT3G02300 | 431 |
| 90 | MA_34295 | AT1G43580 | 378 |
| 91 | MA_30194 | AT5G16210 | 369 |
| 92 | MA_29076 | AT3G57910 | 513 |
| 93 | MA_26068 | AT2G37560 | 414 |
| 94 | MA_25177 | AT1G07970 | 472 |
| 95 | MA_24252 | AT4G24090 | 600 |
| 96 | MA_19954 | AT2G02590 | 414 |
| 97 | MA_11407 | AT3G47860 | 312 |
| 98 | MA_10909 | AT2G04270 | 318 |
| 99 | MA_6888 | AT3G24080 | 2286 |
| 100 | MA_4586 | AT2G22650 | 303 |
| Families | N | Average Locus Recovery | Min | Max |
|---|---|---|---|---|
| Araucariaceae | 5 | 85 | 53 | 97 |
| Cupressaceae | 22 | 98 | 89 | 100 |
| Pinaceae | 11 | 96 | 95 | 90 |
| Podocarpaceae | 26 | 93 | 76 | 97 |
| Sciadopityaceae | 1 | 95 | - | - |
| Taxaceae | 3 | 97 | 96 | 97 |
| Non conifer Gymnosperms | 4 | 81 | 75 | 92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, R.; Biffin, E.; van Dijk, K.-j.; Hill, R.S.; Liu, J.; Waycott, M. Development of a Target Enrichment Probe Set for Conifer (REMcon). Biology 2024, 13, 361. https://doi.org/10.3390/biology13060361
Khan R, Biffin E, van Dijk K-j, Hill RS, Liu J, Waycott M. Development of a Target Enrichment Probe Set for Conifer (REMcon). Biology. 2024; 13(6):361. https://doi.org/10.3390/biology13060361
Chicago/Turabian StyleKhan, Raees, Ed Biffin, Kor-jent van Dijk, Robert S. Hill, Jie Liu, and Michelle Waycott. 2024. "Development of a Target Enrichment Probe Set for Conifer (REMcon)" Biology 13, no. 6: 361. https://doi.org/10.3390/biology13060361