
From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders



{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
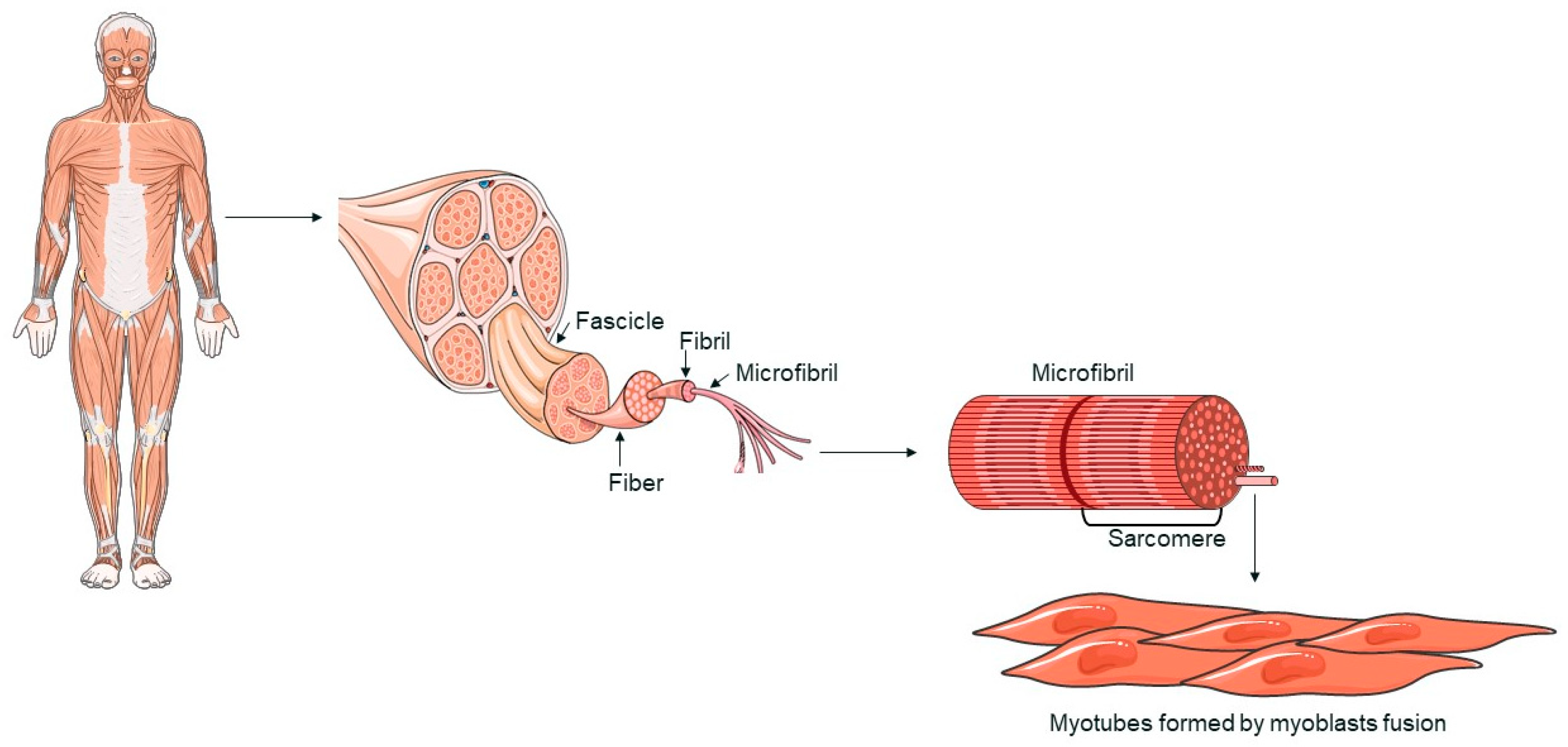
2. An Overview of Muscle Tissue: Structure and Function
3. The Role of Muscle Pathology in ALS: Mechanisms and Treatment Strategies
3.1. Overview of Pathogenic Mechanisms in ALS
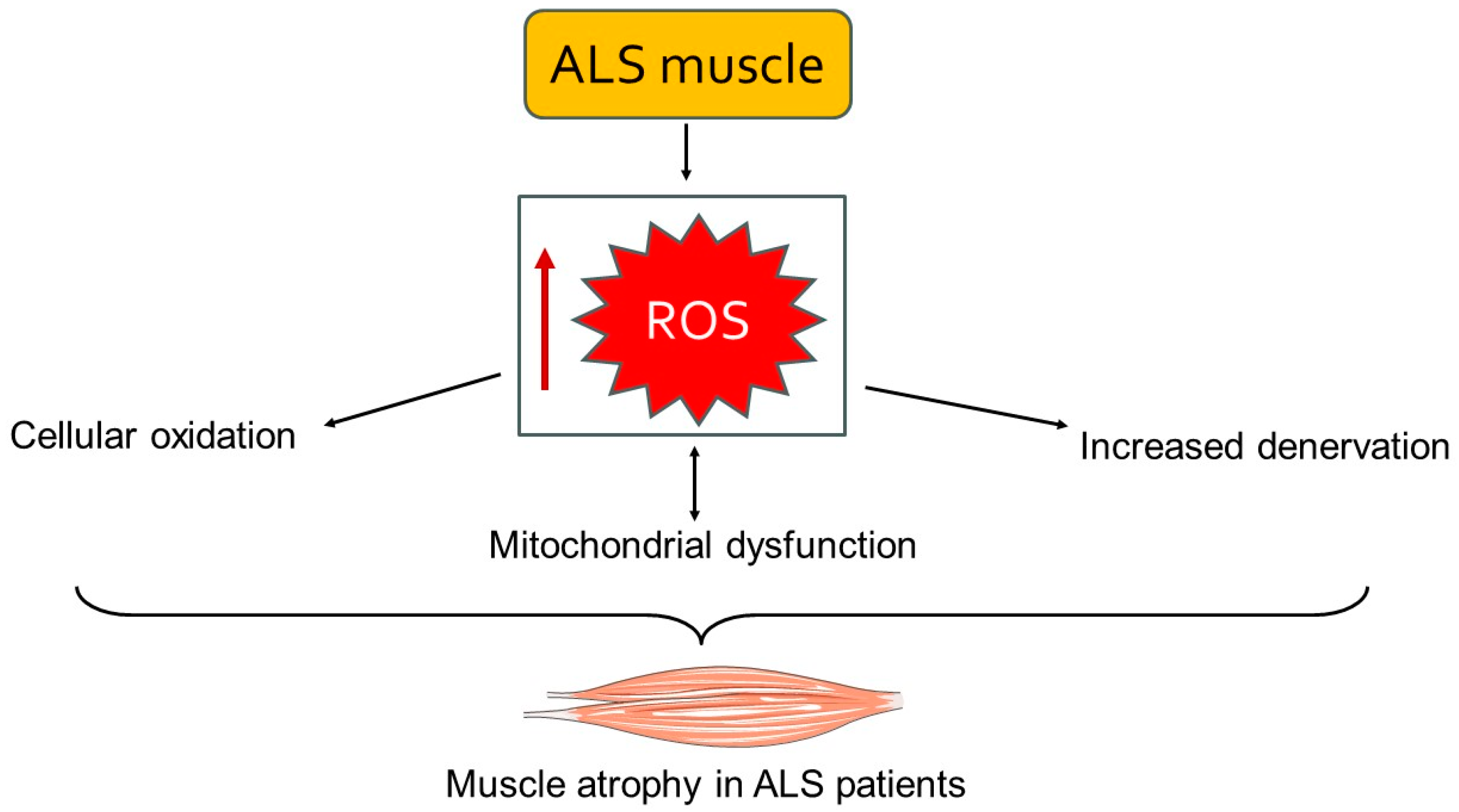
3.2. Involvement of Muscle Tissue in ALS Pathology
3.3. Therapeutic Strategies for Muscle Tissue in ALS
3.3.1. Pharmacological Treatments
3.3.2. Therapeutic Potential of Exercise for Muscle Preservation in ALS
4. Exploring Muscle Pathology in Alzheimer’s Disease: Current Research and Future Directions
4.1. Overview of Pathogenic Mechanisms in AD
4.2. The Impact of Muscle Tissue on AD Pathology
4.3. Therapeutic Approaches for Muscle Tissue in AD
4.3.1. Pharmacological Treatments
4.3.2. Therapeutic Potential of Exercise for Muscle Preservation in AD
5. Muscle Dysfunctions in PD: Implications for Motor Symptoms and Therapeutic Strategies
5.1. Overview of Pathogenic Mechanisms in PD
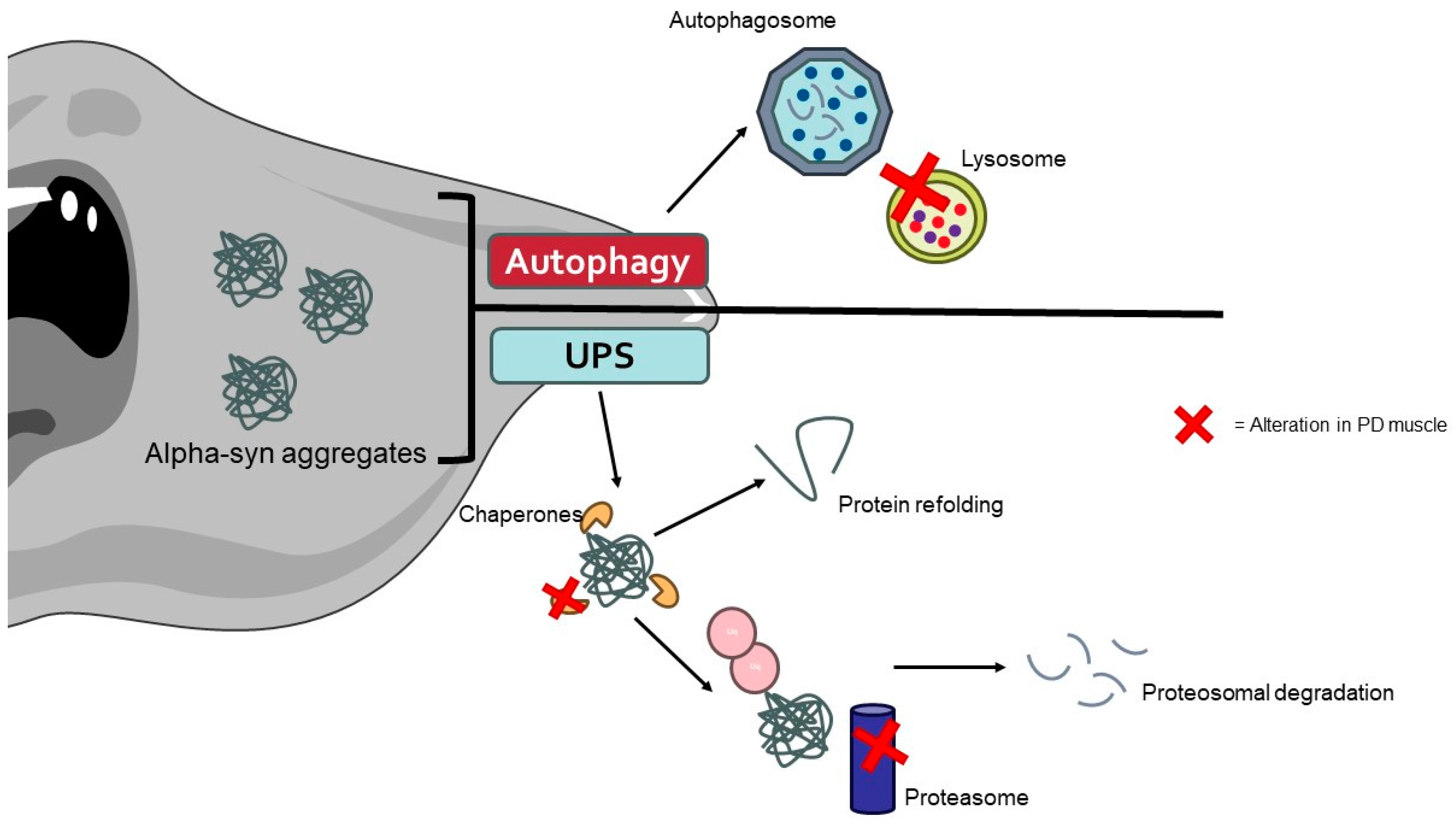
5.2. The Role of Muscle Tissue on PD Pathology
5.3. Therapeutic Interventions for Muscle Tissue in PD
5.3.1. Pharmacological Treatments
5.3.2. Therapeutic Potential of Exercise for Muscle Preservation in PD
6. Muscle Degeneration across Neurodegenerative Disorders: Comparative Insights from ALS, AD, and PD
6.1. Similarities in the Effects on Muscle Tissue
6.1.1. Muscle Atrophy and Weakness
6.1.2. Mitochondrial Dysfunction
6.1.3. Protein Aggregation
6.1.4. Inflammatory Process
6.2. Specific Differences between the Diseases
6.2.1. Mechanisms of Motor Neuron Degeneration
6.2.2. Disease Onset and Progression
6.2.3. Therapeutic Responses
6.2.4. Molecular Pathways
6.2.5. Role of Different Quality Control Mechanisms
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Villa, C.; Paudel, Y.N.; Piperi, C. New Insights into Molecular Mechanisms Underlying Neurodegenerative Disorders. Brain Sci. 2022, 12, 1190. [Google Scholar]
- Duranti, E.; Villa, C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 24, 704. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Muscle Involvement in Amyotrophic Lateral Sclerosis: Understanding the Pathogenesis and Advancing Therapeutics. Biomolecules 2023, 13, 1582. [Google Scholar] [CrossRef] [PubMed]
- Bastin, C.; Giacomelli, F.; Miévis, F.; Lemaire, C.; Guillaume, B.; Salmon, E. Anosognosia in Mild Cognitive Impairment: Lack of Awareness of Memory Difficulties Characterizes Prodromal Alzheimer’s Disease. Front. Psychiatry 2021, 12, 631518. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Váradi, C. Clinical Features of Parkinson’s Disease: The Evolution of Critical Symptoms. Biology 2020, 9, 103. [Google Scholar] [CrossRef]
- Palanisamy, C.P.; Pei, J.; Alugoju, P.; Anthikapalli, N.V.A.; Jayaraman, S.; Veeraraghavan, V.P.; Gopathy, S.; Roy, J.R.; Janaki, C.S.; Thalamati, D.; et al. New strategies of neurodegenerative disease treatment with extracellular vesicles (EVs) derived from mesenchymal stem cells (MSCs). Theranostics 2023, 13, 4138–4165. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Klenerman, D.; Rubinsztein, D.C. Developing Therapies for Neurodegenerative Disorders: Insights from Protein Aggregation and Cellular Stress Responses. Annu. Rev. Cell Dev. Biol. 2020, 36, 165–189. [Google Scholar] [CrossRef]
- Schaffert, L.N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. Int. J. Mol. Sci. 2021, 22, 6016. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Insights into Dysregulated Neurological Biomarkers in Cancer. Cancers 2024, 16, 2680. [Google Scholar] [CrossRef] [PubMed]
- Myszczynska, M.A.; Ojamies, P.N.; Lacoste, A.M.B.; Neil, D.; Saffari, A.; Mead, R.; Hautbergue, G.M.; Holbrook, J.D.; Ferraiuolo, L. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 2020, 16, 440–456. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.; Pinto, M.; Sousa, E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Millet, G.P.; Place, N.; Kayser, B.; Zanou, N. The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. Int. J. Mol. Sci. 2021, 22, 6479. [Google Scholar] [CrossRef]
- Bian, X.; Wang, Q.; Wang, Y.; Lou, S. The function of previously unappreciated exerkines secreted by muscle in regulation of neurodegenerative diseases. Front. Mol. Neurosci. 2023, 16, 1305208. [Google Scholar] [CrossRef]
- Shefner, J.M.; Musaro, A.; Ngo, S.T.; Lunetta, C.; Steyn, F.J.; Robitaille, R.; De Carvalho, M.; Rutkove, S.; Ludolph, A.C.; Dupuis, L. Skeletal muscle in amyotrophic lateral sclerosis. Brain 2023, 146, 4425–4436. [Google Scholar] [CrossRef]
- Beeri, M.S.; Leugrans, S.E.; Delbono, O.; Bennett, D.A.; Buchman, A.S. Sarcopenia is associated with incident Alzheimer’s dementia, mild cognitive impairment, and cognitive decline. J. Am. Geriatr. Soc. 2021, 69, 1826–1835. [Google Scholar] [CrossRef]
- Raleigh, S.M.; Orchard, K.J.A. Sarcopenia as a Risk Factor for Alzheimer’s Disease: Genetic and Epigenetic Perspectives. Genes 2024, 15, 561. [Google Scholar] [CrossRef]
- Halli-Tierney, A.D.; Luker, J.; Carroll, D.G. Parkinson Disease. Am. Fam. Physician 2020, 102, 679–691. [Google Scholar]
- Ferreira-Sánchez, M.D.R.; Moreno-Verdú, M.; Cano-de-la-Cuerda, R. Quantitative Measurement of Rigidity in Parkinson’s Disease: A Systematic Review. Sensors 2020, 20, 880. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.L.; Baumgartner, R.N.; Garry, P.J.; Vellas, B. Advantages of dietary, exercise-related, and therapeutic interventions to prevent and treat sarcopenia in adult patients: An update. Clin. Interv. Aging 2010, 5, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian-Moghadam, H.; Sadat-Shirazi, M.S.; Zarrindast, M.R. Therapeutic potential of stem cells for treatment of neurodegenerative diseases. Biotechnol. Lett. 2020, 42, 1073–1101. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Francaux, M.; Deldicque, L. Exercise and the control of muscle mass in human. Pflug. Arch. 2019, 471, 397–411. [Google Scholar] [CrossRef]
- Rasmussen, B.B.; Phillips, S.M. Contractile and nutritional regulation of human muscle growth. Exerc. Sport. Sci. Rev. 2003, 31, 127–131. [Google Scholar] [CrossRef]
- Westerblad, H.; Bruton, J.D.; Katz, A. Skeletal muscle: Energy metabolism, fiber types, fatigue and adaptability. Exp. Cell Res. 2010, 316, 3093–3099. [Google Scholar] [CrossRef]
- Zurlo, F.; Nemeth, P.M.; Choksi, R.M.; Sesodia, S.; Ravussin, E. Whole-body energy metabolism and skeletal muscle biochemical characteristics. Metabolism 1994, 43, 481–486. [Google Scholar] [CrossRef]
- Kamei, Y.; Hatazawa, Y.; Uchitomi, R.; Yoshimura, R.; Miura, S. Regulation of Skeletal Muscle Function by Amino Acids. Nutrients 2020, 12, 261. [Google Scholar] [CrossRef]
- Fukada, S.I. The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J. Biochem. 2018, 163, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Duranti, E.; Villa, C. Influence of DUX4 Expression in Facioscapulohumeral Muscular Dystrophy and Possible Treatments. Int. J. Mol. Sci. 2023, 24, 9503. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Partridge, T. Skeletal muscle in health and disease. Dis. Models Mech. 2020, 13, dmm042192. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, T.; Toyono, T.; Inoue, A.; Matsubara, T.; Kawamoto, T.; Kokabu, S. Factors Regulating or Regulated by Myogenic Regulatory Factors in Skeletal Muscle Stem Cells. Cells 2022, 11, 1493. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Cretoiu, D.; Pavelescu, L.; Duica, F.; Radu, M.; Suciu, N.; Cretoiu, S.M. Myofibers. Adv. Exp. Med. Biol. 2018, 1088, 23–46. [Google Scholar] [CrossRef]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef]
- Wang, H.; Guan, L.; Deng, M. Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front. Neurosci. 2023, 17, 1170996. [Google Scholar] [CrossRef]
- Irwin, K.E.; Sheth, U.; Wong, P.C.; Gendron, T.F. Fluid biomarkers for amyotrophic lateral sclerosis: A review. Mol. Neurodegener. 2024, 19, 9. [Google Scholar] [CrossRef]
- Kubat, G.B.; Picone, P. Skeletal muscle dysfunction in amyotrophic lateral sclerosis: A mitochondrial perspective and therapeutic approaches. Neurol. Sci. 2024, 45, 4121–4131. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Borasio, G.D. Amyotrophic lateral sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Motataianu, A.; Serban, G.; Barcutean, L.; Balasa, R. Oxidative Stress in Amyotrophic Lateral Sclerosis: Synergy of Genetic and Environmental Factors. Int. J. Mol. Sci. 2022, 23, 9339. [Google Scholar] [CrossRef] [PubMed]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 10, 527. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carrì, M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef]
- Arnold, F.J.; Putka, A.F.; Raychaudhuri, U.; Hsu, S.; Bedlack, R.S.; Bennett, C.L.; La Spada, A.R. Revisiting Glutamate Excitotoxicity in Amyotrophic Lateral Sclerosis and Age-Related Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 5587. [Google Scholar] [CrossRef]
- Li, Q.; Haney, M.S. The role of glia in protein aggregation. Neurobiol. Dis. 2020, 143, 105015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of Neuroinflammation in Amyotrophic Lateral Sclerosis: Cellular Mechanisms and Therapeutic Implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Montiel, M.J.; Chaineau, M.; Durcan, T.M. The Neglected Genes of ALS: Cytoskeletal Dynamics Impact Synaptic Degeneration in ALS. Front. Cell. Neurosci. 2020, 14, 594975. [Google Scholar] [CrossRef]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- Pikatza-Menoio, O.; Elicegui, A.; Bengoetxea, X.; Naldaiz-Gastesi, N.; López de Munain, A.; Gerenu, G.; Gil-Bea, F.J.; Alonso-Martín, S. The Skeletal Muscle Emerges as a New Disease Target in Amyotrophic Lateral Sclerosis. J. Pers. Med. 2021, 11, 671. [Google Scholar] [CrossRef] [PubMed]
- Biedasek, K.; Andres, J.; Mai, K.; Adams, S.; Spuler, S.; Fielitz, J.; Spranger, J. Skeletal muscle 11beta-HSD1 controls glucocorticoid-induced proteolysis and expression of E3 ubiquitin ligases atrogin-1 and MuRF-1. PLoS ONE 2011, 6, e16674. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Cleveland, M.; Pradat, P.F.; Corse, A.; Rothstein, J.D.; Leigh, P.N.; Abila, B.; Bates, S.; Wurthner, J.; et al. Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol. Commun. 2014, 2, 165. [Google Scholar] [CrossRef]
- Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Cicardi, M.E.; Giorgetti, E.; Onesto, E.; Messi, E.; Poletti, A. ALS-related misfolded protein management in motor neurons and muscle cells. Neurochem. Int. 2014, 79, 70–78. [Google Scholar] [CrossRef]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef]
- Colasuonno, F.; Price, R.; Moreno, S. Upper and Lower Motor Neurons and the Skeletal Muscle: Implication for Amyotrophic Lateral Sclerosis (ALS). In Roles of Skeletal Muscle in Organ Development—Prenatal Interdependence among Cells, Tissues, and Organs; Kablar, B., Ed.; Springer: Cham, Switzerland, 2023; Volume 236, pp. 111–129. [Google Scholar] [CrossRef]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Fleming, A.; Bourdenx, M.; Fujimaki, M.; Karabiyik, C.; Krause, G.J.; Lopez, A.; Martín-Segura, A.; Puri, C.; Scrivo, A.; Skidmore, J.; et al. The different autophagy degradation pathways and neurodegeneration. Neuron 2022, 110, 935–966. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.G.; Wehling, S.; Blottner, D. Cell death and apoptosis-related proteins in muscle biopsies of sporadic amyotrophic lateral sclerosis and polyneuropathy. Muscle Nerve 2001, 24, 1083–1089. [Google Scholar] [CrossRef]
- Manzano, R.; Toivonen, J.M.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; López-Royo, T.; Molina, N.; Zaragoza, P.; Calvo, A.C.; Osta, R. What skeletal muscle has to say in amyotrophic lateral sclerosis: Implications for therapy. Br. J. Pharmacol. 2021, 178, 1279–1297. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, C.; Yi, J.; Wu, S.; Luo, G.; Xu, X.; Lin, P.H.; Sun, J.; Zhou, J. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol. Rep. 2015, 3, e12271. [Google Scholar] [CrossRef]
- Chen, W.; Guo, L.; Li, M.; Wei, C.; Li, S.; Xu, R. The pathogenesis of amyotrophic lateral sclerosis: Mitochondrial dysfunction, protein misfolding and epigenetics. Brain Res. 2022, 1786, 147904. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.; Alix, J.J.P.; Neuwirth, C.; Barkhaus, P.E.; Castro, J.; Jenkins, T.M.; McDermott, C.J.; Shaw, P.J.; de Carvalho, M.; Nandedkar, S.; et al. Reinnervation as measured by the motor unit size index is associated with preservation of muscle strength in amyotrophic lateral sclerosis, but not all muscles reinnervate. Muscle Nerve 2022, 65, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, P.A.; Skov, M.; Riisager, A.; Nielsen, O.B.; Pedersen, T.H. Alterations in fast-twitch muscle membrane conductance regulation do not explain decreased muscle function of SOD1(G93A) rats. Muscle Nerve 2021, 64, 755–764. [Google Scholar] [CrossRef]
- Quessada, C.; Bouscary, A.; René, F.; Valle, C.; Ferri, A.; Ngo, S.T.; Loeffler, J.P. Skeletal Muscle Metabolism: Origin or Prognostic Factor for Amyotrophic Lateral Sclerosis (ALS) Development? Cells 2021, 10, 1449. [Google Scholar] [CrossRef]
- Anderson, G. Amyotrophic Lateral Sclerosis Pathoetiology and Pathophysiology: Roles of Astrocytes, Gut Microbiome, and Muscle Interactions via the Mitochondrial Melatonergic Pathway, with Disruption by Glyphosate-Based Herbicides. Int. J. Mol. Sci. 2022, 24, 587. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Scalcon, V.; Massimino, M.L.; Nies, K.; Lopreiato, R.; Rigobello, M.P.; Bertoli, A. SOD1 in ALS: Taking Stock in Pathogenic Mechanisms and the Role of Glial and Muscle Cells. Antioxidants 2022, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Loeffler, J.P. Neuromuscular junction destruction during amyotrophic lateral sclerosis: Insights from transgenic models. Curr. Opin. Pharmacol. 2009, 9, 341–346. [Google Scholar] [CrossRef]
- Lloyd, E.M.; Pinniger, G.J.; Murphy, R.M.; Grounds, M.D. Slow or fast: Implications of myofibre type and associated differences for manifestation of neuromuscular disorders. Acta Physiol. 2023, 238, e14012. [Google Scholar] [CrossRef]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Meo, S. The Role of Reactive Oxygen Species in the Life Cycle of the Mitochondrion. Int. J. Mol. Sci. 2020, 21, 2173. [Google Scholar] [CrossRef]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef]
- Lu, C.H.; Allen, K.; Oei, F.; Leoni, E.; Kuhle, J.; Tree, T.; Fratta, P.; Sharma, N.; Sidle, K.; Howard, R.; et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e244. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. The Role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Jensen, L.; Jørgensen, L.H.; Bech, R.D.; Frandsen, U.; Schrøder, H.D. Skeletal Muscle Remodelling as a Function of Disease Progression in Amyotrophic Lateral Sclerosis. Biomed. Res. Int. 2016, 2016, 5930621. [Google Scholar] [CrossRef]
- Liu, W.; Chakkalakal, J.V. The Composition, Development, and Regeneration of Neuromuscular Junctions. Curr. Top. Dev. Biol. 2018, 126, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Martineau, É.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. eLife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Lepore, E.; Casola, I.; Dobrowolny, G.; Musarò, A. Neuromuscular Junction as an Entity of Nerve-Muscle Communication. Cells 2019, 8, 906. [Google Scholar] [CrossRef]
- Le Grand, F.; Rudnicki, M. Satellite and stem cells in muscle growth and repair. Development 2007, 134, 3953–3957. [Google Scholar] [CrossRef]
- Lynch, K. Optimizing pharmacologic treatment for ALS to improve outcomes and quality of life. Am. J. Manag. Care 2023, 29, S112–S119. [Google Scholar] [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 61–70. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, N.; Nakashima, H.; Fehlings, M.G. Riluzole as a neuroprotective drug for spinal cord injury: From bench to bedside. Molecules 2015, 20, 7775–7789. [Google Scholar] [CrossRef]
- Storch, A.; Burkhardt, K.; Ludolph, A.C.; Schwarz, J. Protective effects of riluzole on dopamine neurons: Involvement of oxidative stress and cellular energy metabolism. J. Neurochem. 2000, 75, 2259–2269. [Google Scholar] [CrossRef]
- Duranti, E.; Cordani, N.; Villa, C. Edaravone: A Novel Possible Drug for Cancer Treatment? Int. J. Mol. Sci. 2024, 25, 1633. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Kim, K. Effects of the Edaravone, a Drug Approved for the Treatment of Amyotrophic Lateral Sclerosis, on Mitochondrial Function and Neuroprotection. Antioxidants 2022, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Morén, C.; deSouza, R.M.; Giraldo, D.M.; Uff, C. Antioxidant Therapeutic Strategies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 9328. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yuki, S.; Watanabe, T.; Mitsuka, M.; Saito, K.I.; Kogure, K. Delayed neuronal death prevented by inhibition of increased hydroxyl radical formation in a transient cerebral ischemia. Brain Res. 1997, 762, 240–242. [Google Scholar] [CrossRef]
- Tzeplaeff, L.; Wilfling, S.; Requardt, M.V.; Herdick, M. Current State and Future Directions in the Therapy of ALS. Cells 2023, 12, 1523. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, Y.; Deng, M. New developments and opportunities in drugs being trialed for amyotrophic lateral sclerosis from 2020 to 2022. Front. Pharmacol. 2022, 13, 1054006. [Google Scholar] [CrossRef]
- Khalaf, K.; Tornese, P.; Cocco, A.; Albanese, A. Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 33. [Google Scholar] [CrossRef]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Nikitin, D.; Makam, A.N.; Suh, K.; McKenna, A.; Carlson, J.J.; Richardson, M.; Rind, D.M.; Pearson, S.D. The effectiveness and value of AMX0035 and oral edaravone for amyotrophic lateral sclerosis: A summary from the Institute for Clinical and Economic Review’s Midwest Comparative Effectiveness Public Advisory Council. J. Manag. Care Spec. Pharm. 2023, 29, 216–221. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Oliveira Santos, M.; de Carvalho, M. Profiling tofersen as a treatment of superoxide dismutase 1 amyotrophic lateral sclerosis. Expert. Rev. Neurother. 2024, 24, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Chawla, P.A. Breaking barriers with tofersen: Enhancing therapeutic opportunities in amyotrophic lateral sclerosis. Eur. J. Neurol. 2024, 31, e16140. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Gillon, A.; Nielsen, K.; Steel, C.; Cornwall, J.; Sheard, P. Exercise attenuates age-associated changes in motoneuron number, nucleocytoplasmic transport proteins and neuromuscular health. Geroscience 2018, 40, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef]
- Sailani, M.R.; Halling, J.F.; Møller, H.D.; Lee, H.; Plomgaard, P.; Pilegaard, H.; Snyder, M.P.; Regenberg, B. Lifelong physical activity is associated with promoter hypomethylation of genes involved in metabolism, myogenesis, contractile properties and oxidative stress resistance in aged human skeletal muscle. Sci. Rep. 2019, 9, 3272. [Google Scholar] [CrossRef]
- Gallo, V.; Vanacore, N.; Bueno-de-Mesquita, H.B.; Vermeulen, R.; Brayne, C.; Pearce, N.; Wark, P.A.; Ward, H.A.; Ferrari, P.; Jenab, M.; et al. Physical activity and risk of Amyotrophic Lateral Sclerosis in a prospective cohort study. Eur. J. Epidemiol. 2016, 31, 255–266. [Google Scholar] [CrossRef]
- Carreras, I.; Yuruker, S.; Aytan, N.; Hossain, L.; Choi, J.K.; Jenkins, B.G.; Kowall, N.W.; Dedeoglu, A. Moderate exercise delays the motor performance decline in a transgenic model of ALS. Brain Res. 2010, 1313, 192–201. [Google Scholar] [CrossRef]
- Tseng, C.; Sinha, K.; Pan, H.; Cui, Y.; Guo, P.; Lin, C.Y.; Yang, F.; Deng, Z.; Eltzschig, H.K.; Lu, A.; et al. Markers of Accelerated Skeletal Muscle Regenerative Response in Murphy Roths Large Mice: Characteristics of Muscle Progenitor Cells and Circulating Factors. Stem Cells 2019, 37, 357–367. [Google Scholar] [CrossRef]
- Bennett, E.J.; Mead, R.J.; Azzouz, M.; Shaw, P.J.; Grierson, A.J. Early detection of motor dysfunction in the SOD1G93A mouse model of Amyotrophic Lateral Sclerosis (ALS) using home cage running wheels. PLoS ONE 2014, 9, e107918. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, J.; Noakes, P.G.; Bellingham, M.C. The Role of Altered BDNF/TrkB Signaling in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2019, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rakotoarisoa, M.; Angelov, B.; Deng, Y.; Angelova, A. Self-Assembled Nanoscale Materials for Neuronal Regeneration: A Focus on BDNF Protein and Nucleic Acid Biotherapeutic Delivery. Nanomaterials 2022, 12, 2267. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Kwak, S.G.; Park, J.S.; Choo, Y.J.; Chang, M.C. Can Therapeutic Exercise Slow Down Progressive Functional Decline in Patients With Amyotrophic Lateral Sclerosis? A Meta-Analysis. Front. Neurol. 2020, 11, 853. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: Results of a randomized, double-blind, controlled study. Sci. Rep. 2019, 9, 2837. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76 Pt A, 27–50. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Villa, C. Biomarkers for Alzheimer’s Disease: Where Do We Stand and Where Are We Going? J. Pers. Med. 2020, 10, 238. [Google Scholar] [CrossRef]
- Ratan, Y.; Rajput, A.; Maleysm, S.; Pareek, A.; Jain, V.; Kaur, R.; Singh, G. An Insight into Cellular and Molecular Mechanisms Underlying the Pathogenesis of Neurodegeneration in Alzheimer’s Disease. Biomedicines 2023, 11, 1398. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Sarnataro, D. New Insights into the Molecular Bases of Familial Alzheimer’s Disease. J. Pers. Med. 2020, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Ryman, D.C.; Acosta-Baena, N.; Aisen, P.S.; Bird, T.; Danek, A.; Fox, N.C.; Goate, A.; Frommelt, P.; Ghetti, B.; Langbaum, J.B.; et al. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology 2014, 83, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Kalaria, R.N.; Cras, P.; Siedlak, S.L.; Velasco, M.E.; Shelton, E.R.; Chan, H.W.; Greenberg, B.D.; Perry, G. Degeneration of vascular muscle cells in cerebral amyloid angiopathy of Alzheimer disease. Brain Res. 1993, 623, 142–146. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kaneko, Y.; Sato, T.; Shimizu, S.; Kanetaka, H.; Hanyu, H. Sarcopenia and Muscle Functions at Various Stages of Alzheimer Disease. Front. Neurol. 2018, 9, 710. [Google Scholar] [CrossRef]
- Boyle, P.A.; Buchman, A.S.; Wilson, R.S.; Leurgans, S.E.; Bennett, D.A. Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch. Neurol. 2009, 66, 1339–1344. [Google Scholar] [CrossRef]
- Mani, S.; Dubey, R.; Lai, I.C.; Babu, M.A.; Tyagi, S.; Swargiary, G.; Mody, D.; Singh, M.; Agarwal, S.; Iqbal, D.; et al. Oxidative Stress and Natural Antioxidants: Back and Forth in the Neurological Mechanisms of Alzheimer’s Disease. J. Alzheimers Dis. 2023, 96, 877–912. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Tian, Q.; Bilgel, M.; Walker, K.A.; Moghekar, A.R.; Fishbein, K.W.; Spencer, R.G.; Resnick, S.M.; Ferrucci, L. Skeletal muscle mitochondrial function predicts cognitive impairment and is associated with biomarkers of Alzheimer’s disease and neurodegeneration. Alzheimers Dement. 2023, 19, 4436–4445. [Google Scholar] [CrossRef] [PubMed]
- Turkseven, C.H.; Buyukakilli, B.; Balli, E.; Yetkin, D.; Erdal, M.E.; Yilmaz, S.G.; Sahin, L. Effects of Huperzin-A on the Beta-amyloid accumulation in the brain and skeletal muscle cells of a rat model for Alzheimer’s disease. Life Sci. 2017, 184, 47–57. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Yamazaki, T.; Lemere, C.A.; Frosch, M.P.; Selkoe, D.J. Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease. An immunoelectron microscopic study. Am. J. Pathol. 1992, 141, 249–259. [Google Scholar] [PubMed]
- Sehar, U.; Rawat, P.; Reddy, A.P.; Kopel, J.; Reddy, P.H. Amyloid Beta in Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 12924. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef]
- Thakur, S.; Dhapola, R.; Sarma, P.; Medhi, B.; Reddy, D.H. Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Marino, M.; Scuderi, F.; Provenzano, C.; Bartoccioni, E. Skeletal muscle cells: From local inflammatory response to active immunity. Gene Ther. 2011, 18, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Oudbier, S.J.; Goh, J.; Looijaard, S.; Reijnierse, E.M.; Meskers, C.G.M.; Maier, A.B. Pathophysiological Mechanisms Explaining the Association Between Low Skeletal Muscle Mass and Cognitive Function. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1959–1968. [Google Scholar] [CrossRef]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and Insulin Resistance in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Chakraborty, P.; Bhattacharya, H.; Chacko, L.; Singh, B.; Chaudhary, A.; Javvaji, K.; Pradhan, S.R.; Vallamkondu, J.; Dey, A.; et al. Altered glucose metabolism in Alzheimer’s disease: Role of mitochondrial dysfunction and oxidative stress. Free Radic. Biol. Med. 2022, 193, 134–157. [Google Scholar] [CrossRef]
- Ezkurdia, A.; Ramírez, M.J.; Solas, M. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance. Int. J. Mol. Sci. 2023, 24, 4354. [Google Scholar] [CrossRef]
- Arosio, B.; Calvani, R.; Ferri, E.; Coelho-Junior, H.J.; Carandina, A.; Campanelli, F.; Ghiglieri, V.; Marzetti, E.; Picca, A. Sarcopenia and Cognitive Decline in Older Adults: Targeting the Muscle-Brain Axis. Nutrients 2023, 15, 1853. [Google Scholar] [CrossRef] [PubMed]
- Filardi, M.; Barone, R.; Bramato, G.; Nigro, S.; Tafuri, B.; Frisullo, M.E.; Zecca, C.; Tortelli, R.; Logroscino, G. The Relationship between Muscle Strength and Cognitive Performance across Alzheimer’s Disease Clinical Continuum. Front. Neurol. 2022, 13, 833087. [Google Scholar] [CrossRef]
- Sugimoto, T.; Ono, R.; Murata, S.; Saji, N.; Matsui, Y.; Niida, S.; Toba, K.; Sakurai, T. Prevalence and associated factors of sarcopenia in elderly subjects with amnestic mild cognitive impairment or Alzheimer disease. Curr. Alzheimer Res. 2016, 13, 718–726. [Google Scholar] [CrossRef]
- Beckett, T.L.; Niedowicz, D.M.; Studzinski, C.M.; Weidner, A.M.; Webb, R.L.; Holler, C.J.; Ahmed, R.R.; LeVine, H., 3rd; Murphy, M.P. Effects of nonsteroidal anti-inflammatory drugs on amyloid-beta pathology in mouse skeletal muscle. Neurobiol. Dis. 2010, 39, 449–456. [Google Scholar] [CrossRef]
- Alturki, M.; Beyer, I.; Mets, T.; Bautmans, I. Impact of drugs with anti-inflammatory effects on skeletal muscle and inflammation: A systematic literature review. Exp. Gerontol. 2018, 114, 33–49. [Google Scholar] [CrossRef]
- Mantle, D.; Heaton, R.A.; Hargreaves, I.P. Coenzyme Q10, Ageing and the Nervous System: An Overview. Antioxidants 2021, 11, 2. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Arenas-de Larriva, A.P.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q(10) Supplementation for the Reduction of Oxidative Stress: Clinical Implications in the Treatment of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed]
- Vints, W.A.J.; Levin, O.; van Griensven, M.; Vlaeyen, J.W.S.; Masiulis, N.; Verbunt, J.; van Laake-Geelen, C.C.M. Neuromuscular electrical stimulation to combat cognitive aging in people with spinal cord injury: Protocol for a single case experimental design study. BMC Neurol. 2024, 24, 197. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Ahmad, K.; Lee, E.J.; Lee, Y.H.; Choi, I. Implications of Insulin-Like Growth Factor-1 in Skeletal Muscle and Various Diseases. Cells 2020, 9, 1773. [Google Scholar] [CrossRef]
- Zemva, J.; Schubert, M. The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: Possible therapeutic implications. CNS Neurol. Disord. Drug Targets 2014, 13, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Van Broeck, B.; Van Broeckhoven, C.; Kumar-Singh, S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener. Dis. 2007, 4, 349–365. [Google Scholar] [CrossRef]
- Franco-Bocanegra, D.K.; McAuley, C.; Nicoll, J.A.R.; Boche, D. Molecular Mechanisms of Microglial Motility: Changes in Ageing and Alzheimer’s Disease. Cells 2019, 8, 639. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef] [PubMed]
- Tanganelli, F.; Meinke, P.; Hofmeister, F.; Jarmusch, S.; Baber, L.; Mehaffey, S.; Hintze, S.; Ferrari, U.; Neuerburg, C.; Kammerlander, C.; et al. Type-2 muscle fiber atrophy is associated with sarcopenia in elderly men with hip fracture. Exp. Gerontol. 2021, 144, 111171. [Google Scholar] [CrossRef]
- Moon, Y.; Choi, Y.J.; Kim, J.O.; Han, S.H. Muscle profile and cognition in patients with Alzheimer’s disease dementia. Neurol. Sci. 2018, 39, 1861–1866. [Google Scholar] [CrossRef]
- Veronese, N.; Solmi, M.; Basso, C.; Smith, L.; Soysal, P. Role of physical activity in ameliorating neuropsychiatric symptoms in Alzheimer disease: A narrative review. Int. J. Geriatr. Psychiatry 2019, 34, 1316–1325. [Google Scholar] [CrossRef]
- Santos-Lozano, A.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Quindós-Rubial, M.; Fiuza-Luces, C.; Cristi-Montero, C.; Emanuele, E.; Garatachea, N.; Lucia, A. Physical Activity and Alzheimer Disease: A Protective Association. Mayo Clin. Proc. 2016, 91, 999–1020. [Google Scholar] [CrossRef] [PubMed]
- Brasure, M.; Desai, P.; Davila, H.; Nelson, V.A.; Calvert, C.; Jutkowitz, E.; Butler, M.; Fink, H.A.; Ratner, E.; Hemmy, L.S.; et al. Physical Activity Interventions in Preventing Cognitive Decline and Alzheimer-Type Dementia: A Systematic Review. Ann. Intern. Med. 2018, 168, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Holsinger, R.M.D. Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Ageing Res. Rev. 2018, 48, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.; Cipriano, I.; Costa, T.; Saraiva, M.; Martins, A. Exercise, ageing and cognitive function—Effects of a personalized physical exercise program in the cognitive function of older adults. Physiol. Behav. 2019, 202, 8–13. [Google Scholar] [CrossRef]
- Polito, R.; Di Meo, I.; Barbieri, M.; Daniele, A.; Paolisso, G.; Rizzo, M.R. Adiponectin Role in Neurodegenerative Diseases: Focus on Nutrition Review. Int. J. Mol. Sci. 2020, 21, 9255. [Google Scholar] [CrossRef]
- Dvorak, R.V.; Poehlman, E.T. Appendicular skeletal muscle mass, physical activity, and cognitive status in patients with Alzheimer’s disease. Neurology 1998, 51, 1386–1390. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Q.; Quan, H.; Kang, S.G.; Huang, K.; Tong, T. Nutraceuticals in the Prevention and Treatment of the Muscle Atrophy. Nutrients 2021, 13, 1914. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Thomas, C.J.; Radcliffe, J.; Itsiopoulos, C. Omega-3 Fatty Acids in Early Prevention of Inflammatory Neurodegenerative Disease: A Focus on Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 172801. [Google Scholar] [CrossRef]
- Nolan, J.M.; Power, R.; Howard, A.N.; Bergin, P.; Roche, W.; Prado-Cabrero, A.; Pope, G.; Cooke, J.; Power, T.; Mulcahy, R. Supplementation With Carotenoids, Omega-3 Fatty Acids, and Vitamin E Has a Positive Effect on the Symptoms and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2022, 90, 233–249. [Google Scholar] [CrossRef]
- De Sousa, O.V.; Mendes, J.; Amaral, T.F. Nutritional and Functional Indicators and Their Association With Mortality Among Older Adults With Alzheimer’s Disease. Am. J. Alzheimers Dis. Other Dement. 2020, 35, 1533317520907168. [Google Scholar] [CrossRef]
- Feraco, P.; Gagliardo, C.; La Tona, G.; Bruno, E.; D’Angelo, C.; Marrale, M.; Del Poggio, A.; Malaguti, M.C.; Geraci, L.; Baschi, R.; et al. Imaging of Substantia Nigra in Parkinson’s Disease: A Narrative Review. Brain Sci. 2021, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Vélez, G.E.; Zoghbi, H.Y. Parkinson’s Disease Genetics and Pathophysiology. Annu. Rev. Neurosci. 2021, 44, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Goyal, L.; Singh, S. Tremor and Rigidity in Patients with Parkinson’s Disease: Emphasis on Epidemiology, Pathophysiology and Contributing Factors. CNS Neurol. Disord. Drug Targets 2022, 21, 596–609. [Google Scholar] [CrossRef]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Bougea, A.; Papageorgiou, S.G.; Villa, C. Psychosis in Parkinson’s Disease: A Lesson from Genetics. Genes 2022, 13, 1099. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef]
- Ajith, C.; Divya, K.P.; Asish, V. Parkinson’s disease—Genetic cause. Curr. Opin. Neurol. 2023, 36, 292–301. [Google Scholar] [CrossRef]
- Menšíková, K.; Matěj, R.; Colosimo, C.; Rosales, R.; Tučková, L.; Ehrmann, J.; Hraboš, D.; Kolaříková, K.; Vodička, R.; Vrtěl, R.; et al. Lewy body disease or diseases with Lewy bodies? npj Park. Dis. 2022, 8, 3. [Google Scholar] [CrossRef]
- Schulz-Schaeffer, W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010, 120, 131–143. [Google Scholar] [CrossRef]
- Jęśko, H.; Lenkiewicz, A.M.; Wilkaniec, A.; Adamczyk, A. The interplay between parkin and alpha-synuclein; possible implications for the pathogenesis of Parkinson’s disease. Acta Neurobiol. Exp. 2019, 79, 276–289. [Google Scholar] [CrossRef]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New Perspectives on Roles of Alpha-Synuclein in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 370. [Google Scholar] [CrossRef] [PubMed]
- Moradi Vastegani, S.; Nasrolahi, A.; Ghaderi, S.; Belali, R.; Rashno, M.; Farzaneh, M.; Khoshnam, S.E. Mitochondrial Dysfunction and Parkinson’s Disease: Pathogenesis and Therapeutic Strategies. Neurochem. Res. 2023, 48, 2285–2308. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Dadarao Chakole, R.; Nemade, L.S.; Kishor Kale, N.; Borah, S.; Shrikant Deokar, S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis—An updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Park. Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef]
- Zuo, L.; Motherwell, M.S. The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 2013, 532, 18–23. [Google Scholar] [CrossRef]
- Oczkowska, A.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson’s Disease. Curr. Genom. 2013, 14, 502–517. [Google Scholar] [CrossRef]
- Lotankar, S.; Prabhavalkar, K.S.; Bhatt, L.K. Biomarkers for Parkinson’s Disease: Recent Advancement. Neurosci. Bull. 2017, 33, 585–597. [Google Scholar] [CrossRef]
- Karabiyik, C.; Lee, M.J.; Rubinsztein, D.C. Autophagy impairment in Parkinson’s disease. Essays Biochem. 2017, 61, 711–720. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Behl, T.; Kumar, S.; Althafar, Z.M.; Sehgal, A.; Singh, S.; Sharma, N.; Badavath, V.N.; Yadav, S.; Bhatia, S.; Al-Harrasi, A.; et al. Exploring the Role of Ubiquitin-Proteasome System in Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 4257–4273. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, G.; Ren, M.; Sun, T.; Li, Y.; Ye, M.; Ma, C.; Guo, Y.; Liu, C. The Role of Ubiquitin-Proteasome System and Mitophagy in the Pathogenesis of Parkinson’s Disease. Neuromol. Med. 2023, 25, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the Role of Neuroinflammation in the Progression of Parkinson’s Disease. Front. Neurol. 2018, 9, 860. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharm. Res. 2019, 42, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef]
- Surendranathan, A.; Su, L.; Mak, E.; Passamonti, L.; Hong, Y.T.; Arnold, R.; Vázquez Rodríguez, P.; Bevan-Jones, W.R.; Brain, S.A.E.; Fryer, T.D.; et al. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 2018, 141, 3415–3427. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Lavin, K.M.; Ge, Y.; Sealfon, S.C.; Nair, V.D.; Wilk, K.; McAdam, J.S.; Windham, S.T.; Kumar, P.L.; McDonald, M.N.; Bamman, M.M. Rehabilitative Impact of Exercise Training on Human Skeletal Muscle Transcriptional Programs in Parkinson’s Disease. Front. Physiol. 2020, 11, 653. [Google Scholar] [CrossRef]
- Kaufmann, H.; Goldstein, D.S. Autonomic dysfunction in Parkinson disease. Handb. Clin. Neurol. 2013, 117, 259–278. [Google Scholar] [CrossRef]
- Gdynia, H.J.; Sperfeld, A.D.; Unrath, A.; Ludolph, A.C.; Sabolek, M.; Storch, A.; Kassubek, J. Histopathological analysis of skeletal muscle in patients with Parkinson’s disease and ‘dropped head’/’bent spine’ syndrome. Park. Relat. Disord. 2009, 15, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Gamborg, M.; Hvid, L.G.; Thrue, C.; Johansson, S.; Franzén, E.; Dalgas, U.; Langeskov-Christensen, M. Muscle Strength and Power in People With Parkinson Disease: A Systematic Review and Meta-analysis. J. Neurol. Phys. Ther. 2023, 47, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.M.; Roberts, T.; Hodder, J.; Allen, P.S.; Zhu, G.; Martin, W.R. Generalized mitochondrial dysfunction in Parkinson’s disease detected by magnetic resonance spectroscopy of muscle. Neurology 1995, 45, 2097–2099. [Google Scholar] [CrossRef]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar] [CrossRef]
- Schulz, J.B.; Beal, M.F. Mitochondrial dysfunction in movement disorders. Curr. Opin. Neurol. 1994, 7, 333–339. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.; Zhao, C.; Pang, S.; Lu, J.; Chan, P. α-Synuclein aggregation causes muscle atrophy through neuromuscular junction degeneration. J. Cachexia Sarcopenia Muscle 2023, 14, 226–242. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, Y.; Zhang, Y. Mitochondrial reactive oxygen species cause major oxidative mitochondrial DNA damages and repair pathways. J. Biosci. 2020, 45, 84. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Zhou, T.T.; Zhang, Y.; Chen, N.H.; Yuan, Y.H. Distribution of α-Synuclein Aggregation in the Peripheral Tissues. Neurochem. Res. 2022, 47, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Padhy, A.A.; Kumari, V.; Mishra, P. Role of Ubiquitin-Proteasome and Autophagy-Lysosome Pathways in α-Synuclein Aggregate Clearance. Mol. Neurobiol. 2022, 59, 5379–5407. [Google Scholar] [CrossRef]
- Le Guerroué, F.; Youle, R.J. Ubiquitin signaling in neurodegenerative diseases: An autophagy and proteasome perspective. Cell Death Differ. 2021, 28, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Sala, G.; Marinig, D.; Arosio, A.; Ferrarese, C. Role of Chaperone-Mediated Autophagy Dysfunctions in the Pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 157. [Google Scholar] [CrossRef]
- Basellini, M.J.; Kothuis, J.M.; Comincini, A.; Pezzoli, G.; Cappelletti, G.; Mazzetti, S. Pathological Pathways and Alpha-Synuclein in Parkinson’s Disease: A View from the Periphery. Front. Biosci. 2023, 28, 33. [Google Scholar] [CrossRef]
- Mu, L.; Sobotka, S.; Chen, J.; Su, H.; Sanders, I.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Samanta, J.E.; Beach, T.G. Alpha-synuclein pathology and axonal degeneration of the peripheral motor nerves innervating pharyngeal muscles in Parkinson disease. J. Neuropathol. Exp. Neurol. 2013, 72, 119–129. [Google Scholar] [CrossRef]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Wang, T.; Shi, C.; Luo, H.; Zheng, H.; Fan, L.; Tang, M.; Su, Y.; Yang, J.; Mao, C.; Xu, Y. Neuroinflammation in Parkinson’s Disease: Triggers, Mechanisms, and Immunotherapies. Neuroscientist 2022, 28, 364–381. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Shen, F.F.; Yuan, Y.S.; Li, X.; Ji, P.; Zhu, L.; Sun, L.; Ding, J.; Niu, Q.; et al. Activated Schwann cells and increased inflammatory cytokines IL-1β, IL-6, and TNF-α in patients’ sural nerve are lack of tight relationship with specific sensory disturbances in Parkinson’s disease. CNS Neurosci. Ther. 2020, 26, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Thundyil, J.; Lim, K.L. DAMPs and neurodegeneration. Ageing Res. Rev. 2015, 24, 17–28. [Google Scholar] [CrossRef]
- Moehlman, A.T.; Kanfer, G.; Youle, R.J. Loss of STING in parkin mutant flies suppresses muscle defects and mitochondria damage. PLoS Genet. 2023, 19, e1010828. [Google Scholar] [CrossRef]
- Ji, Y.; Li, M.; Chang, M.; Liu, R.; Qiu, J.; Wang, K.; Deng, C.; Shen, Y.; Zhu, J.; Wang, W.; et al. Inflammation: Roles in Skeletal Muscle Atrophy. Antioxidants 2022, 11, 1686. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.N. Mitochondrial enhancement for neurodegenerative movement disorders: A systematic review of trials involving creatine, coenzyme Q10, idebenone and mitoquinone. CNS Drugs 2014, 28, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Avila, M.; Vega, A.F.; de la Mata, M.; Pavon, A.D.; Alcocer-Gomez, E.; Calero, C.P.; Paz, M.V.; Alanis, M.; et al. Clinical applications of coenzyme Q10. Front. Biosci. 2014, 19, 619–633. [Google Scholar] [CrossRef]
- Arenas-Jal, M.; Suñé-Negre, J.M.; García-Montoya, E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr. Rev. Food Sci. Food Saf. 2020, 19, 574–594. [Google Scholar] [CrossRef]
- Delgobo, M.; Agnes, J.P.; Gonçalves, R.M.; Dos Santos, V.W.; Parisotto, E.B.; Zamoner, A.; Zanotto-Filho, A. N-acetylcysteine and alpha-lipoic acid improve antioxidant defenses and decrease oxidative stress, inflammation and serum lipid levels in ovariectomized rats via estrogen-independent mechanisms. J. Nutr. Biochem. 2019, 67, 190–200. [Google Scholar] [CrossRef]
- Ciulla, M.; Marinelli, L.; Cacciatore, I.; Stefano, A.D. Role of Dietary Supplements in the Management of Parkinson’s Disease. Biomolecules 2019, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Alrouji, M.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Saad, H.M.; Batiha, G.E. A story of the potential effect of non-steroidal anti-inflammatory drugs (NSAIDs) in Parkinson’s disease: Beneficial or detrimental effects. Inflammopharmacology 2023, 31, 673–688. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. Non-steroidal anti-inflammatory drugs (NSAIDs) and other anti-inflammatory agents in the treatment of neurodegenerative disease. Curr. Alzheimer Res. 2005, 2, 355–365. [Google Scholar] [CrossRef]
- Fouré, A.; Bendahan, D. Is Branched-Chain Amino Acids Supplementation an Efficient Nutritional Strategy to Alleviate Skeletal Muscle Damage? A Systematic Review. Nutrients 2017, 9, 1047. [Google Scholar] [CrossRef]
- Neinast, M.; Murashige, D.; Arany, Z. Branched Chain Amino Acids. Annu. Rev. Physiol. 2019, 81, 139–164. [Google Scholar] [CrossRef]
- Shimomura, Y.; Yamamoto, Y.; Bajotto, G.; Sato, J.; Murakami, T.; Shimomura, N.; Kobayashi, H.; Mawatari, K. Nutraceutical effects of branched-chain amino acids on skeletal muscle. J. Nutr. 2006, 136, 529s–532s. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, R.R. Branched-chain amino acids and muscle protein synthesis in humans: Myth or reality? J. Int. Soc. Sports Nutr. 2017, 14, 30. [Google Scholar] [CrossRef]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yang, F.; Sun, L.; Yu, J.; Si, Y.; Yao, L. Role of gut microbiota-derived branched-chain amino acids in the pathogenesis of Parkinson’s disease: An animal study. Brain Behav. Immun. 2022, 106, 307–321. [Google Scholar] [CrossRef]
- Tosukhowong, P.; Boonla, C.; Dissayabutra, T.; Kaewwilai, L.; Muensri, S.; Chotipanich, C.; Joutsa, J.; Rinne, J.; Bhidayasiri, R. Biochemical and clinical effects of Whey protein supplementation in Parkinson’s disease: A pilot study. J. Neurol. Sci. 2016, 367, 162–170. [Google Scholar] [CrossRef]
- Matthews, V.B.; Aström, M.B.; Chan, M.H.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Akerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef]
- Krumpolec, P.; Vallova, S.; Slobodova, L.; Tirpakova, V.; Vajda, M.; Schon, M.; Klepochova, R.; Janakova, Z.; Straka, I.; Sutovsky, S.; et al. Aerobic-Strength Exercise Improves Metabolism and Clinical State in Parkinson’s Disease Patients. Front. Neurol. 2017, 8, 698. [Google Scholar] [CrossRef] [PubMed]
- Schootemeijer, S.; van der Kolk, N.M.; Bloem, B.R.; de Vries, N.M. Current Perspectives on Aerobic Exercise in People with Parkinson’s Disease. Neurotherapeutics 2020, 17, 1418–1433. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, G.; Rafferty, M.R.; Prodoehl, J.; Kohrt, W.M.; Comella, C.L.; Simuni, T.; Corcos, D.M. Effects of endurance exercise training on the motor and non-motor features of Parkinson’s disease: A review. J. Park. Dis. 2015, 5, 21–41. [Google Scholar] [CrossRef]
- De Oliveira, M.P.B.; Lobato, D.F.M.; Smaili, S.M.; Carvalho, C.; Borges, J.B.C. Effect of aerobic exercise on functional capacity and quality of life in individuals with Parkinson’s disease: A systematic review of randomized controlled trials. Arch. Gerontol. Geriatr. 2021, 95, 104422. [Google Scholar] [CrossRef] [PubMed]
- Supriya, R.; Singh, K.P.; Gao, Y.; Gu, Y.; Baker, J.S. Effect of Exercise on Secondary Sarcopenia: A Comprehensive Literature Review. Biology 2021, 11, 51. [Google Scholar] [CrossRef]
- Silva-Batista, C.; Mattos, E.C.; Corcos, D.M.; Wilson, J.M.; Heckman, C.J.; Kanegusuku, H.; Piemonte, M.E.; Túlio de Mello, M.; Forjaz, C.; Roschel, H.; et al. Resistance training with instability is more effective than resistance training in improving spinal inhibitory mechanisms in Parkinson’s disease. J. Appl. Physiol. 2017, 122, 1–10. [Google Scholar] [CrossRef]
- Silva-Batista, C.; Corcos, D.M.; Kanegusuku, H.; Piemonte, M.E.P.; Gobbi, L.T.B.; de Lima-Pardini, A.C.; de Mello, M.T.; Forjaz, C.L.M.; Ugrinowitsch, C. Balance and fear of falling in subjects with Parkinson’s disease is improved after exercises with motor complexity. Gait Posture 2018, 61, 90–97. [Google Scholar] [CrossRef]
- Elangovan, N.; Cheung, C.; Mahnan, A.; Wyman, J.F.; Tuite, P.; Konczak, J. Hatha yoga training improves standing balance but not gait in Parkinson’s disease. Sports Med. Health Sci. 2020, 2, 80–88. [Google Scholar] [CrossRef]
- Feng, Y.S.; Yang, S.D.; Tan, Z.X.; Wang, M.M.; Xing, Y.; Dong, F.; Zhang, F. The benefits and mechanisms of exercise training for Parkinson’s disease. Life Sci. 2020, 245, 117345. [Google Scholar] [CrossRef]
- Hackney, M.E.; Wolf, S.L. Impact of Tai Chi Chu’an practice on balance and mobility in older adults: An integrative review of 20 years of research. J. Geriatr. Phys. Ther. 2014, 37, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, S.; Hennlein, L.; Sendtner, M. Therapy development for spinal muscular atrophy: Perspectives for muscular dystrophies and neurodegenerative disorders. Neurol. Res. Pract. 2022, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Tiryaki, E.; Horak, H.A. ALS and other motor neuron diseases. Continuum 2014, 20, 1185–1207. [Google Scholar] [CrossRef]
- Uitti, R.J.; Berry, K.; Yasuhara, O.; Eisen, A.; Feldman, H.; McGeer, P.L.; Calne, D.B. Neurodegenerative ‘overlap’ syndrome: Clinical and pathological features of Parkinson’s disease, motor neuron disease, and Alzheimer’s disease. Park. Relat. Disord. 1995, 1, 21–34. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Duarte, A.I.; Santos, M.S.; Rego, A.C.; Oliveira, C.R. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 741–761. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007, 14, 1261–1266. [Google Scholar] [CrossRef]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein aggregation in the brain: The molecular basis for Alzheimer’s and Parkinson’s diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef]
- Abyadeh, M.; Gupta, V.; Paulo, J.A.; Mahmoudabad, A.G.; Shadfar, S.; Mirshahvaladi, S.; Nguyen, C.T.O.; Finkelstein, D.I.; You, Y.; Haynes, P.A.; et al. Amyloid-beta and tau protein beyond Alzheimer’s disease. Neural Regen. Res. 2024, 19, 1262–1276. [Google Scholar] [CrossRef]
- Khan, M.S.H.; Hegde, V. Obesity and Diabetes Mediated Chronic Inflammation: A Potential Biomarker in Alzheimer’s Disease. J. Pers. Med. 2020, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Inflammatory process in Parkinson’s disease: Role for cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Park. Relat. Disord. 2012, 18 (Suppl. S1), S207–S209. [Google Scholar] [CrossRef] [PubMed]
- Choe, M.A.; Koo, B.S.; An, G.J.; Jeon, S. Effects of Treadmill Exercise on the Recovery of Dopaminergic Neuron Loss and Muscle Atrophy in the 6-OHDA Lesioned Parkinson’s Disease Rat Model. Korean J. Physiol. Pharmacol. 2012, 16, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.S.; Bennett, D.A. Loss of motor function in preclinical Alzheimer’s disease. Expert. Rev. Neurother. 2011, 11, 665–676. [Google Scholar] [CrossRef]
- Martignon, C.; Ruzzante, F.; Giuriato, G.; Laginestra, F.G.; Pedrinolla, A.; Di Vico, I.A.; Saggin, P.; Stefanelli, D.; Tinazzi, M.; Schena, F.; et al. The key role of physical activity against the neuromuscular deterioration in patients with Parkinson’s disease. Acta Physiol. 2021, 231, e13630. [Google Scholar] [CrossRef]
- Bettio, L.E.B.; Rajendran, L.; Gil-Mohapel, J. The effects of aging in the hippocampus and cognitive decline. Neurosci. Biobehav. Rev. 2017, 79, 66–86. [Google Scholar] [CrossRef]
- Archer, T. Physical exercise alleviates debilities of normal aging and Alzheimer’s disease. Acta Neurol. Scand. 2011, 123, 221–238. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The Role of Exercise as a Non-pharmacological Therapeutic Approach for Amyotrophic Lateral Sclerosis: Beneficial or Detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef]
- Lautenschlager, N.T.; Cox, K.L.; Ellis, K.A. Physical activity for cognitive health: What advice can we give to older adults with subjective cognitive decline and mild cognitive impairment? Dialogues Clin. Neurosci. 2019, 21, 61–68. [Google Scholar] [CrossRef]
- Pham, J.; Keon, M.; Brennan, S.; Saksena, N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. Int. J. Mol. Sci. 2020, 21, 3464. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Chen, S.; Xu, C. Functions of amyloid precursor protein in metabolic diseases. Metabolism 2021, 115, 154454. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervous system: Implications for neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duranti, E.; Villa, C. From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders. Biology 2024, 13, 719. https://doi.org/10.3390/biology13090719
Duranti E, Villa C. From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders. Biology. 2024; 13(9):719. https://doi.org/10.3390/biology13090719
Chicago/Turabian StyleDuranti, Elisa, and Chiara Villa. 2024. "From Brain to Muscle: The Role of Muscle Tissue in Neurodegenerative Disorders" Biology 13, no. 9: 719. https://doi.org/10.3390/biology13090719