
ITS2 rRNA Gene Sequence–Structure Phylogeny of the Chytridiomycota (Opisthokonta, Fungi)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
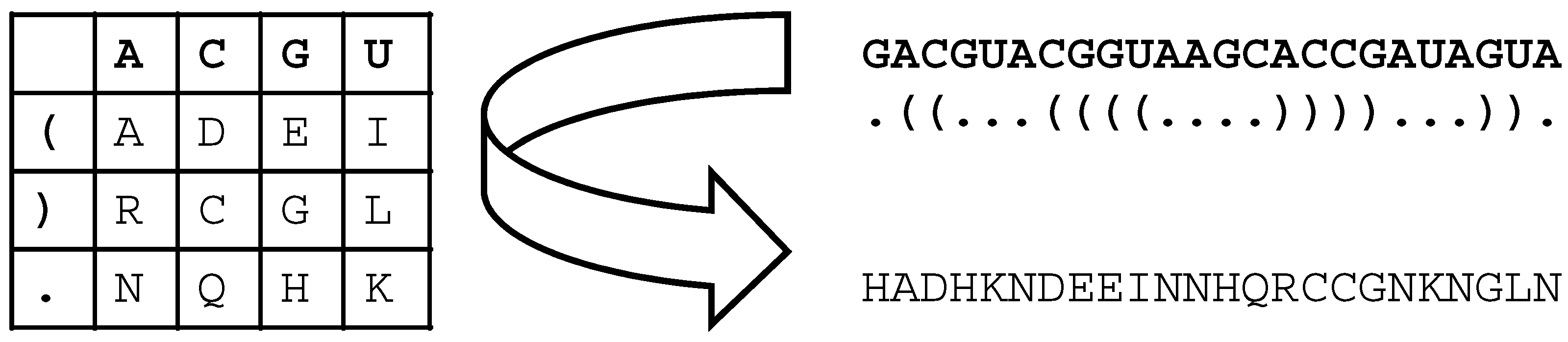
2. Materials and Methods
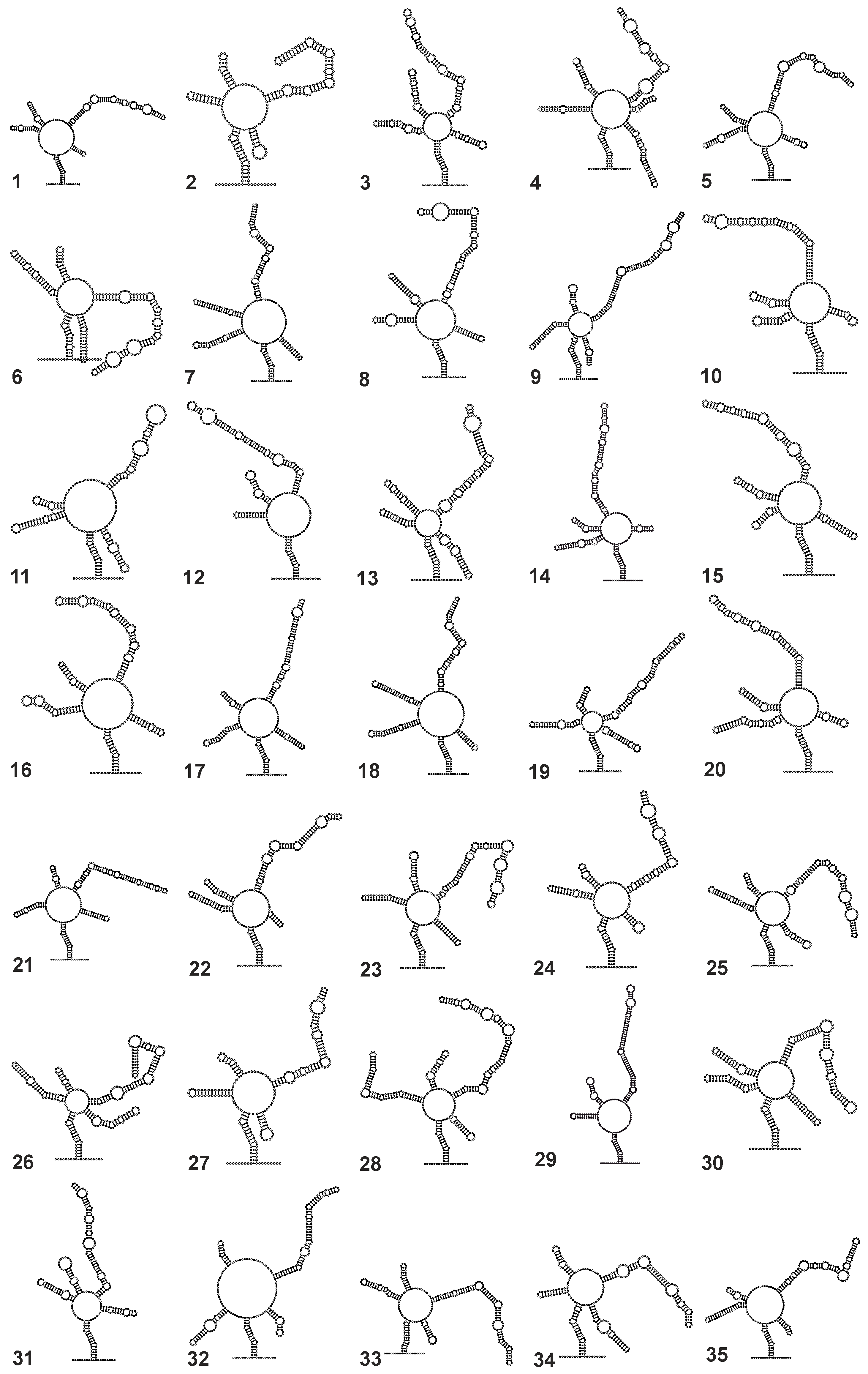
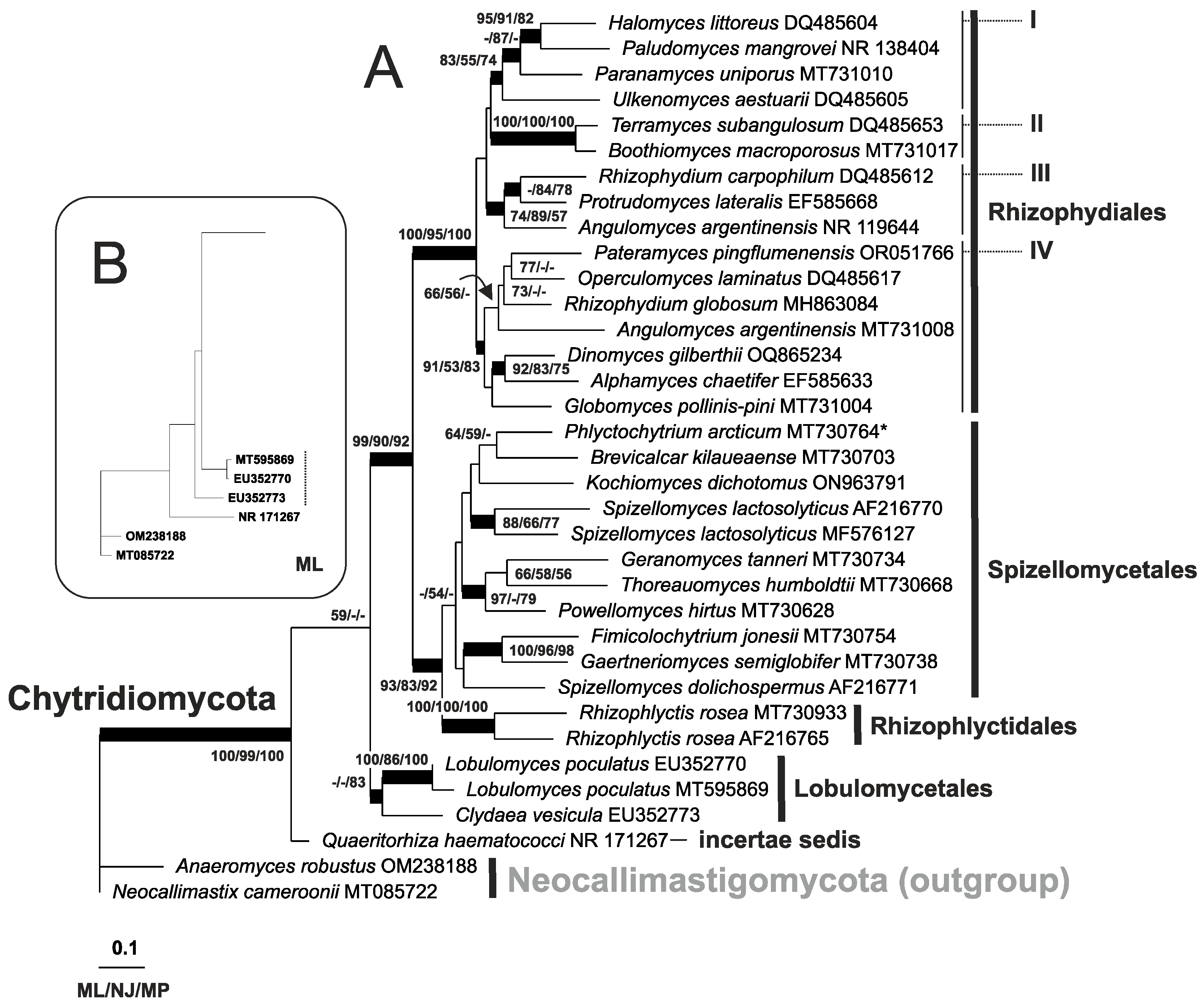
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powell, M.J.; Letcher, P.M. Chytridiomycota, Monoblepharidomycota, and Neocallimastigomycota. In Systematics and Evolution; McLaughlin, D.J., Spatafora, J.W., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 141–175. [Google Scholar]
- Powell, M.J.; Letcher, P.M. From zoospores to molecules: The evolution and systematics of Chytridiomycota. In Systematics and Evolution of Fungi; Misra, J.K., Tewari, J.P., Deshmukh, S.K., Eds.; CRC Press: New York, NY, USA, 2012; pp. 29–54. [Google Scholar]
- Letcher, P.M.; Powell, M.J. Hypothesized evolutionary trends in zoospore ultrastructural characters in Chytridiales (Chytridiomycota). Mycologia 2014, 106, 379–396. [Google Scholar] [CrossRef]
- James, T.Y.; Letcher, P.M.; Longcore, J.E.; Mozley-Standridge, S.E.; Porter, D.; Powell, M.J.; Griffith, G.W.; Vilgalys, R. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 2006, 98, 860–871. [Google Scholar] [CrossRef] [PubMed]
- James, T.Y.; Kauff, F.; Schoch, C.L.; Matheny, P.B.; Hofstetter, V.; Cox, C.J.; Celio, G.; Gueidan, C.; Fraker, E.; Miadlikowska, J.; et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 2006, 443, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hibbett, D.S.; Binder, M.; Bischoff, J.F.; Blackwell, M.; Cannon, P.F.; Eriksson, O.E.; Huhndorf, S.; James, T.; Kirk, P.M.; Lücking, R.; et al. A higher-level phylogenetic classification of the Fungi. Mycol. Res. 2007, 111, 509–547. [Google Scholar] [CrossRef] [PubMed]
- Barr, D. Phylum Chytridiomycota. In Handbook of Protoctista; Margulis, L., Corliss, J.O., Melkonian, M., Chapman, D.J., Eds.; Jones & Bartlett Boston: Boston, MA, USA, 1990. [Google Scholar]
- Powell, M.J.; Letcher, P.M. Phylogeny and characterization of freshwater Chytridiomycota (Chytridiomycetes and Monoblepharidomycetes). In Freshwater Fungi; Jones, E.B.G., Kevin, D.H., Ka-Lai, P., Eds.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2014; pp. 133–154. [Google Scholar]
- Wijayawardene, N.N.; Pawłowska, J.; Letcher, P.M.; Kirk, P.M.; Humber, R.A.; Schüßler, A.; Wrzosek, M.; Muszewska, A.; Okrasińska, A.; Istel, Ł.; et al. Notes for genera: Basal clades of Fungi (including Aphelidiomycota, Basidiobolomycota, Blastocladiomycota, Calcarisporiellomycota, Caulochytriomycota, Chytridiomycota, Entomophthoromycota, Glomeromycota, Kickxellomycota, Monoblepharomycota, Mortierellomyc. Fungal Divers. 2018, 92, 43–129. [Google Scholar] [CrossRef]
- Seto, K.; Van Den Wyngaert, S.; Degawa, Y.; Kagami, M. Taxonomic revision of the genus Zygorhizidium: Zygorhizidiales and Zygophlyctidales ord. nov.(Chytridiomycetes, Chytridiomycota). Fungal Syst. Evol. 2020, 5, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.; Kagami, M.; Degawa, Y. Phylogenetic Position of Parasitic Chytrids on Diatoms: Characterization of a Novel Clade in Chytridiomycota. J. Eukaryot. Microbiol. 2017, 64, 383–393. [Google Scholar] [CrossRef]
- Letcher, P.M.; Powell, M.J.; Churchill, P.F.; Chambers, J.G. Ultrastructural and molecular phylogenetic delineation of a new order, the Rhizophydiales (Chytridiomycota). Mycol. Res. 2006, 110, 898–915. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B.; List, F.B.C.A.; Bolchacova, E. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef]
- Keller, A.; Schleicher, T.; Schultz, J.; Müller, T.; Dandekar, T.; Wolf, M. 5.8S-28S rRNA interaction and HMM-based ITS2 annotation. Gene 2009, 430, 50–57. [Google Scholar] [CrossRef]
- Keller, A.; Förster, F.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. Including RNA secondary structures improves accuracy and robustness in reconstruction of phylogenetic trees. Biol. Direct 2010, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Koetschan, C.; Müller, T. ITS2, 18S, 16S or any other RNA—simply aligning sequences and their individual secondary structures simultaneously by an automatic approach. Gene 2014, 546, 145–149. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau Donald, C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 50, D20–D26. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Ankenbrand, M.J.; Keller, A.; Wolf, M.; Schultz, J.; Förster, F. ITS2 Database V: Twice as Much. Mol. Biol. Evol. 2015, 32, 3030–3032. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Wolf, M. Synchronous visual analysis and editing of RNA sequence and secondary structure alignments using 4SALE. BMC Res. Notes 2008, 1, 91. [Google Scholar] [CrossRef] [PubMed]
- Seibel, P.N.; Müller, T.; Dandekar, T.; Schultz, J.; Wolf, M. 4SALE—A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform. 2006, 7, 498. [Google Scholar] [CrossRef]
- Selig, C.; Wolf, M.; Muller, T.; Dandekar, T.; Schultz, J. The ITS2 Database II: Homology modelling RNA structure for molecular systematics. Nucleic Acids Res. 2007, 36, D377–D380. [Google Scholar] [CrossRef] [PubMed]
- Hepperle, D. Align Ver. 07/04. Multisequence Alignment Editor and Preparation/Manipulation of Phylogenetic Datasets. Available online: https://www.sequentix.de/ (accessed on 19 September 2024).
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Camin, J.H.; Sokal, R.R. A Method for Deducing Branching Sequences in Phylogeny. Evolution 1965, 19, 311–326. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Ruderisch, B.; Dandekar, T.; Schultz, J.; Müller, T. ProfDistS: (profile-) distance based phylogeny on sequence—Structure alignments. Bioinformatics 2008, 24, 2401–2402. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D. PAUP*. Phylogenetic Analysis Using Parsimony (*And Other Methods), Version 4.0b10; Sinauer Associates: Sunderland, UK, 2002.
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org (accessed on 19 September 2024).
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Barr, D.J.S. An outline for the reclassification of the Chytridiales, and for a new order, the Spizellomycetales. Can. J. Bot. 1980, 58, 2380–2394. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalaiventhan, Y.; Seto, K.; Wolf, M. ITS2 rRNA Gene Sequence–Structure Phylogeny of the Chytridiomycota (Opisthokonta, Fungi). Biology 2025, 14, 36. https://doi.org/10.3390/biology14010036
Kalaiventhan Y, Seto K, Wolf M. ITS2 rRNA Gene Sequence–Structure Phylogeny of the Chytridiomycota (Opisthokonta, Fungi). Biology. 2025; 14(1):36. https://doi.org/10.3390/biology14010036
Chicago/Turabian StyleKalaiventhan, Yuveantheni, Kensuke Seto, and Matthias Wolf. 2025. "ITS2 rRNA Gene Sequence–Structure Phylogeny of the Chytridiomycota (Opisthokonta, Fungi)" Biology 14, no. 1: 36. https://doi.org/10.3390/biology14010036
APA StyleKalaiventhan, Y., Seto, K., & Wolf, M. (2025). ITS2 rRNA Gene Sequence–Structure Phylogeny of the Chytridiomycota (Opisthokonta, Fungi). Biology, 14(1), 36. https://doi.org/10.3390/biology14010036