Simple Summary
Interleukin-23 is a crucial protein produced by the immune system that plays a role in triggering and developing certain inflammatory diseases. It stimulates specific immune cells, like Th17 cells, which are involved in inflammatory diseases including psoriasis, rheumatoid arthritis, inflammatory bowel disease, and multiple sclerosis. This review examines how IL-23 contributes to these diseases and discusses the potential of treatments that target IL-23 and its receptor.
Abstract
Interleukin-23 is crucial in the initiation and progression of certain inflammatory disorders. As a key cytokine, IL-23 is involved in the differentiation and activation of Th17 cells, which play a role in a broad spectrum of inflammatory diseases. This review examines the molecular mechanisms through which IL-23 contributes to the pathogenesis of conditions including psoriasis, rheumatoid arthritis, inflammatory bowel disease, and multiple sclerosis. By elucidating the significant role of IL-23 in inflammation, this review underscores its importance as a therapeutic target for managing inflammatory conditions, with particular emphasis on current and emerging biologic treatments.
1. Introduction
Inflammation is a fundamental biological process that plays a critical role in immune defense and the maintenance of tissue homeostasis [1]. This highly regulated and dynamic response functions as a protective mechanism, enabling an organism to detect and eliminate harmful agents, such as pathogens, apoptotic and necrotic cells, and toxic substances [2]. Under physiological conditions, the inflammatory response is controlled to ensure effective immune function while minimizing tissue damage. Once the harmful stimulus is eliminated, resolution mechanisms actively restore homeostasis, preventing unnecessary tissue injury [3].
At the molecular level, inflammation is initiated by the activation of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs), upon the recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [4]. This activation triggers intracellular signaling cascades, such as the NF-κB transcription factor and MAPK pathways, which drive the transcription of key pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 [5]. These cytokines further amplify the inflammatory response by recruiting immune cells, including neutrophils, macrophages, and lymphocytes, to the site of infection or injury [6].
The resolution of inflammation is an active and tightly coordinated process mediated by specialized pro-resolving lipid mediators (SPMs), including resolvins, protectins, and maresins, which are derived from ω-3 polyunsaturated fatty acids [7]. These biomolecules counteract pro-inflammatory signaling, evoke the macrophage-mediated clearance of apoptotic cells (efferocytosis), and inhibit excessive neutrophil infiltration, ultimately restoring tissue integrity and function [8]. The dysregulation of these pathways promotes sustained inflammation and contributes to the development of many chronic inflammatory diseases, in which the immune system erroneously attacks the body’s own tissues [9].
Prolonged and uncontrolled inflammation is strongly linked to several diseases that compromise quality of life [10]. Among these, chronic inflammatory disorders like psoriasis, inflammatory bowel disease (IBD), rheumatoid arthritis (RA), and multiple sclerosis (MS) are the most common and clinically significant manifestations of immune dysregulation [11,12,13]. In these pathological conditions, the dysregulated activation of immune cells, including Th cells, B cells, and antigen-presenting cells (APCs), results in the biosynthesis of pro-inflammatory cytokines such as IFN-γ, IL-17, and GM-CSF [14,15]. These cytokines play a key role in sustaining and amplifying the inflammatory response, creating a vicious cycle in which inflammation persists, leading to tissue damage [16]. Among them, IL-23 has emerged as a critical mediator in the pathogenesis of autoimmune and inflammatory diseases, attracting considerable scientific interest [17].
IL-23 is a heterodimeric cytokine composed of two distinct subunits: p19, which confers its specific biological function [18], and p40, a component it shares with interleukin-12 (IL-12) [19]. It is predominantly produced by several APCs, such as macrophages and dendritic cells, in response to microbial stimuli and other immune signals [20]. Since its discovery, IL-23 has been identified as a regulator of pathogenic inflammation, primarily due to its role in driving the differentiation, expansion, and sustained activation of Th17 cells [21]. Th17 cells secrete a range of pro-inflammatory cytokines, such as IL-17, IL-22, and GM-CSF, which collectively contribute to immune cell activation and subsequent tissue damage [22]. While Th17 cells can fulfill protective and homeostatic functions under certain physiological conditions [23], their dysregulated activation in the presence of IL-23 drives persistent inflammation and progressive tissue destruction.
Given its central role in sustaining chronic inflammation, IL-23 has emerged as a key target for therapeutic intervention. Monoclonal antibodies designed to selectively neutralize IL-23 have demonstrated high efficacy in the treatment of autoimmune diseases, especially psoriasis and IBD [24]. These immunotherapies include ustekinumab, which targets the shared p40 subunit of both IL-12 and IL-23 [25], as well as more selective p19 inhibitors such as guselkumab and risankizumab [26,27]. By disrupting the IL-23-mediated inflammatory cascade, these treatments have yielded many clinical benefits, leading to symptom relief and disease remission in affected individuals.
This review provides a comprehensive evaluation of IL-23 in the onset and progression of inflammatory diseases. By evaluating its immunological functions, role in disease pathogenesis, and therapeutic potential, this paper highlights IL-23 as both a biomarker and a target for therapeutic intervention in many chronic inflammatory diseases. Enhancing our understanding of IL-23 in immune regulation will not only expand our knowledge of the mechanisms underlying inflammatory diseases but also contribute to the creation of more effective and personalized treatment approaches.
2. Biology of IL-23
In this section, the molecular structure of IL-23 as well as its receptor (IL-23R) will be thoroughly analyzed. The structure of IL-23 will be examined, focusing on the molecular interactions (with IL-23R) that allow IL-23 to perform its biological functions. Moreover, this section will explore the cellular actions triggered by the binding of IL-23 with IL-23R. Upon receptor activation, many intracellular signaling pathways are initiated. These signaling events are crucial to the role of IL-23 in immune regulation, particularly in the differentiation of Th17 cells and the subsequent production of pro-inflammatory cytokines. The consequences of the IL-23/IL-23R interaction will be discussed in the context of immune responses and inflammation. Finally, the relevance of IL-23 and IL-23R in the development of autoimmune and inflammatory diseases will be addressed.
2.1. Molecular Structure of IL-23
IL-23 is a heterodimeric cytokine with a molecular weight of approximately 59 kDa, composed of 476 amino acids [19]. It consists of two subunits: IL-23p19 (p19), a subunit unique to IL-23, and IL-12p40 (p40), a subunit shared with IL-12 [18,19]. Beyond its structural similarities with IL-12, IL-23 shares both evolutionary and functional links with other cytokines in the IL-12 family, such as IL-27 and IL-35 [28]. While IL-12 and IL-23 predominantly drive pro-inflammatory immune responses, IL-27 and IL-35 are mainly linked to immunoregulatory functions, underscoring the complex interplay among these cytokines in the maintenance of immune homeostasis [29].
The crystal structure of IL-23 (Figure 1) provides important insights into its molecular architecture and functional interactions. IL-23p19 adopts a characteristic four-helix bundle cytokine fold, a structural motif commonly found in members of the IL-6 family, which facilitates its specific binding interactions [18]. On the other hand, IL-12p40 is organized into three distinct structural domains, forming a scaffold that plays a critical role in receptor engagement and signal transduction [19]. When the two subunits assemble, they form a complex three-dimensional structure that is essential to its biological function. The molecular configuration of IL-23 features a unique conformational arrangement, wherein a key arginine residue (Arg159) on helix D of IL-23p19 interacts with a highly specific binding site on IL-12p40, known as the “arginine pocket” [30]. This interaction is driven by numerous molecular forces, including hydrogen bonding, electrostatic interactions, and van der Waals forces, all of which contribute to the stability and receptor binding capacity of the cytokine [31].
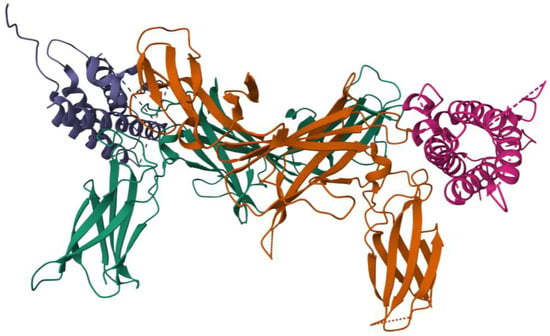
Figure 1.
Three-dimensional structure of IL-23. Image provided by RCSB Protein Data Bank.
Functionally, the p19 subunit is chiefly responsible for binding to the IL-23R complex, a pivotal determinant in the activation of downstream signaling pathways that promote Th17 cell differentiation and pro-inflammatory cytokine production [32]. In contrast, IL-12p40 is essential for mediating interactions between the IL-23 and the IL-12Rβ1, a receptor subunit also employed by IL-12 [31]. This dual-receptor binding characteristic emphasizes the evolutionary and functional relationship between IL-12 and IL-23, highlighting the different shared signaling mechanisms that regulate immune homeostasis and inflammatory responses [28].
2.2. IL-23 Receptor: Structure and Localization
The IL-23R complex is composed of two distinct type I transmembrane proteins: IL-12Rβ1 and IL-23R [31]. While IL-12Rβ1 serves as a shared receptor subunit also employed in the IL-12 receptor complex [33], IL-23R is unique to IL-23 signaling [34]. IL-23R is mainly expressed in several immune cells, playing a key role in mediating inflammatory responses and immune regulation [31]. Notably, its expression is found in macrophages [35], NKT cells [36], ILCs [37], γδ T cells [32], MAITs [38], and Th17 cells [32]. Structurally, IL-23R is a 629-amino acid glycoprotein with a molecular weight of approximately 70 kDa [39]. The receptor is composed of several functional domains, each contributing to its role in ligand binding and intracellular signal transduction. IL-23R contains an N-terminal signal peptide essential for its proper localization to the plasma membrane, a fibronectin type III-like domain that facilitates ligand binding and receptor dimerization, and an intracellular cytoplasmic tail enriched with three conserved tyrosine phosphorylation sites that serve as docking sites for downstream signaling molecules [39].
Upon IL-23 binding (Figure 2), IL-23R begins a complex intracellular signaling cascade mainly mediated by certain members of the JAK family [34]. IL-23R-associated JAK2 and IL-12Rβ1-associated TYK2 undergo activation through trans-phosphorylation events. These kinases subsequently phosphorylate specific tyrosine residues within the cytoplasmic domain of IL-23R, creating docking sites for STAT3 [24,40,41]. Phosphorylated STAT3 undergoes dimerization and subsequent translocation to the nucleus, where it acts as a transcriptional regulator, orchestrating the expression of many genes involved in immune activation [42]. Among these, one of the most critical targets is RORγt, a master transcription factor essential for Th17 cell differentiation [43].
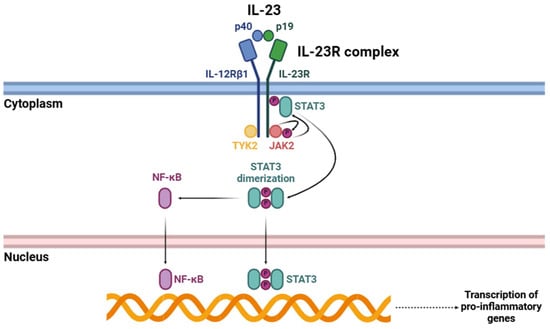
Figure 2.
Signaling cascade triggered by the interaction of IL-23 with its receptor (IL-23R). Abbreviations: p40 (p40 subunit), p19 (p19 subunit), IL-12Rβ1 (interleukin 12 receptor β1), IL-23R (interleukin 23 receptor), STAT3 (signal transducer and activator of transcription 3), TYK2 (tyrosine kinase 2), JAK2 (Janus kinase 2), and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells).
Apart from the well-characterized JAK-STAT signaling pathway, the activation of IL-23R triggers other intracellular signaling pathways that further amplify its pro-inflammatory effects. One of the key pathways triggered by IL-23R is the NF-κB signaling pathway, which is crucial for regulating the transcription of numerous pro-inflammatory cytokines and other immune effector molecules [44,45]. Upon IL-23R activation, structural changes occur that facilitate the recruitment and activation of different intracellular signaling components. This process activates key kinases, including IκB kinase (IKK), which phosphorylates IκB proteins, leading to their degradation [46]. This proteolytic degradation releases NF-κB dimers, primarily p65/p50, allowing their translocation into the nucleus [47]. Once in the nucleus, these NF-κB dimers bind to specific DNA sequences within the promoters of pro-inflammatory genes, thereby triggering their transcription [48]. Consequently, this pathway enhances the production of various cytokines and effector molecules that contribute to the amplification and persistence of inflammatory responses.
Pro-inflammatory cytokines upregulated through the NF-κB signaling pathway include IL-6 [49], IL-17 [50], IL-21 [51], IL-22 [52], GM-CSF [53], and TNF-α [54]. These cytokines play a key role in inflammation by recruiting immune cells, as well as contributing to dysregulated tissue remodeling and maladaptive immune responses linked to chronic inflammatory diseases.
2.3. Biological Roles of IL-23
IL-23 plays a pivotal role in various immune responses, particularly in the regulation of inflammation and host defense mechanisms. This cytokine is synthesized in response to inflammatory stimuli, microbial invasion, or pathogenic insults, which trigger its production by various immune and non-immune cell types [55,56,57]. Among the most significant producers of IL-23 are antigen-presenting cells (APCs), including macrophages, monocytes, and dendritic cells (DCs) [58]. These cells respond to pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), inducing the transcription and secretion of IL-23 [59].
Beyond these primary sources, IL-23 can also be synthesized by innate immune cells like neutrophils and innate lymphoid cells (ILCs), especially ILC3s. Neutrophils, recognized for their role in acute inflammation and pathogen clearance, have been found to contribute to the IL-23-mediated immune response under certain inflammatory conditions [60]. Similarly, ILC3s participate in mucosal immunity by responding to IL-23 signaling, leading to the production of IL-17 and IL-22, cytokines essential for epithelial barrier integrity and antimicrobial defense [61].
In addition to microbial stimuli, IL-23 production is influenced by various environmental and intracellular factors. Hypoxia and metabolic signals have been shown to modulate IL-23 expression, adding another layer of complexity to its regulation [62,63]. Hypoxic conditions, often present in inflamed tissues, enhance IL-23 synthesis via HIF-1α, further amplifying inflammatory responses [62]. Similarly, metabolic signals, such as disruptions in lipid metabolism within macrophages, can drive IL-23 production, linking immune activation to metabolic pathways [63].
In the skin, IL-23 is produced by a range of cell types, including keratinocytes, dermal dendritic cells (dDCs), and epidermal Langerhans cells (Figure 3) [64,65,66]. The cutaneous production of IL-23 is particularly relevant in the context of autoimmune and inflammatory skin disorders, such as psoriasis, where its overexpression contributes to chronic inflammation by promoting the expansion of Th17 cells [21]. Additionally, the gastrointestinal tract serves as another critical site of IL-23 synthesis, where its production is largely driven by Paneth cells (specialized epithelial cells located in the small intestinal crypts) [67]. These cells play an essential role in maintaining mucosal immunity and regulating the intestinal microbiota by releasing antimicrobial peptides in response to bacterial stimuli [68]. Additionally, within the gut-associated lymphoid tissue (GALT), DCs and macrophages produce IL-23, particularly in IBD [69,70].
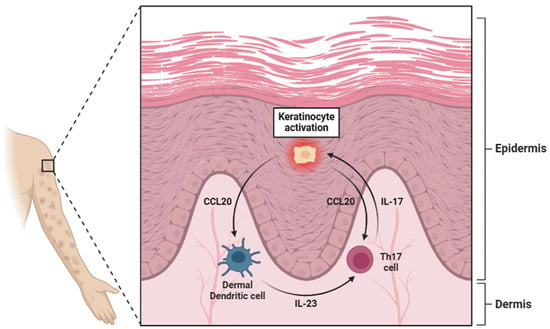
Figure 3.
A diagram illustrating the pivotal role of keratinocytes in psoriasis. These cells activate dermal dendritic cells (dDCs) through the secretion of CCL20, which, through interaction with specific receptors (CCR6), stimulates the dDCs to release IL-23. This cytokine activates Th17 lymphocytes through binding to the IL-23R receptor. Once activated, Th17 cells secrete IL-17, which in turn further activates keratinocytes, reinforcing a positive feedback loop. However, the secretion of CCL20 by keratinocytes also exerts a degree of regulation over Th17 lymphocytes. Abbreviations: CCL20 (C-C motif chemokine ligand 20), IL-23 (interleukin 23), Th17 (T helper 17 cell), and IL-17 (interleukin 17).
The principal biological function of IL-23 lies in its crucial role in sustaining and amplifying the Th17 cell lineage, a subset of CD4+ T cells known for their potent pro-inflammatory activity [71]. Although IL-23 is not directly involved in the initial differentiation of naïve CD4+ T cells into Th17 cells (a process primarily driven by cytokines such as TGF-β and IL-6), it plays an indispensable role in the stabilization, expansion, and maintenance of this population [72,73]. Once Th17 cells have differentiated, IL-23 promotes their survival and proliferation, reinforcing their effector functions and enhancing their capacity to secrete pro-inflammatory cytokines, including IL-17, IL-21, and IL-22 [74]. These cytokines orchestrate a robust inflammatory cascade by activating immune cells at sites of infection or tissue damage [75].
Finally, accumulating evidence suggests that IL-23 plays a multifaceted role in tumor immunity, exerting pro-tumorigenic and anti-tumorigenic effects depending on the tumor microenvironment, immune cell composition, and cytokine signaling dynamics [76]. IL-23 promotes the recruitment and activation of numerous immune effector cells, including CD8+ T cells, NK cells, and neutrophils, within the TME [77]. These immune cells contribute to tumor cell lysis through various mechanisms: (1) IL-23 stimulates Th17 cells to produce IL-17A and IL-22, which enhance the infiltration of effector immune cells into the TME. IL-17A induces the expression of chemokines such as CCL20, CXCL1, and CXCL9, which facilitate the recruitment of DCs, NK cells, and CD8+ T lymphocytes [78,79]. (2) IL-23 promotes the maturation of DCs, enhancing their ability to present tumor antigens to naïve T cells, a crucial process for initiating adaptive immune responses that target tumor cells [80]. (3) IL-23 amplifies antitumor immunity by enhancing the cytotoxic activity of CD8+ T lymphocytes and NK cells, leading to increased production and secretion of perforin and granzyme B, which facilitate tumor cell apoptosis [81].
Paradoxically, IL-23 can also contribute to tumor progression in certain malignancies by creating a pro-inflammatory microenvironment that promotes immune evasion, angiogenesis, and malignant cell survival. This dual function is context-dependent and shaped by the presence of different immunosuppressive cells, cytokine interactions, and metabolic factors within the TME [82]. IL-23, through the action of IL-17, facilitates the recruitment of additional pro-inflammatory cells, including neutrophils and MDSCs [83]. These recruited cells have the ability to inhibit anti-tumor immunity, thereby enhancing the tumor’s capacity to evade immunosurveillance [84]. Simultaneously, IL-23 is implicated in the modulation of angiogenesis, a hallmark of tumor progression [85]. Through the induction of VEGF production (via IL-17), IL-23 indirectly facilitates neovascularization within the tumor, thereby ensuring an adequate supply of nutrients and oxygen to rapidly proliferating neoplastic cells [86]. This angiogenic response not only promotes tumor growth but also plays a role in creating a more intricate and immunosuppressive TME [87].
The impact of IL-23 on immune evasion is further reinforced by its interactions with Tregs. In numerous malignancies, IL-23 can enhance the accumulation of Tregs, which are known to exert immunosuppressive effects within the TME [88]. By increasing the expression of immunosuppressive molecules such as TGF-β and IL-10 [89,90], Tregs induced by IL-23 help to attenuate the activity of CTLs and NKs, thereby facilitating tumor immune escape [91].
These conditions can amplify the pro-tumorigenic effects of IL-23 by driving a metabolic shift that supports tumor cell survival and immune tolerance, thereby establishing a feedback loop that perpetuates the inflammatory microenvironment, which in turn promotes tumor growth.
3. Role of IL-23 in Pain-Associated Inflammatory Mechanisms
One of the most compelling pieces of evidence for the role of IL-23 in inflammatory pain comes from several studies involving genetically modified mice deficient in the IL-23p19 subunit, which is essential for the functional activity of IL-23 [92,93,94,95,96,97]. These studies have demonstrated that the absence of IL-23 exerts a profound protective effect against the development of zymosan-induced arthritis (ZIA), an established preclinical model of RA characterized by robust immune activation and pain-related behaviors [98]. Notably, IL-23p19-deficient mice exhibit a complete abolition of both the pathological hallmarks of ZIA and the behavioral manifestations of pain typically observed in this preclinical model, suggesting that IL-23 is indispensable for the initiation and propagation of inflammation-driven nociceptive signaling [93]. The absence of IL-23, as demonstrated in IL23p19−/− mice, disrupts this intricate network, leading to a marked reduction in the recruitment and activation of immune cells within affected tissues, thereby mitigating pain hypersensitivity and tissue damage [94].
In the context of experimental autoimmune encephalomyelitis (EAE), the most commonly used experimental model for human inflammatory demyelinating disease, including MS [99], IL-23 stimulates myelin-reactive T cells to produce both IFN-γ and IL-17, contributing to the generation of encephalitogenic Th17 cells [100]. The differentiation of naive CD4+ T cells into Th17 effector cells is primarily driven by TGF-β and IL-6, with IL-23 playing a critical role in their survival and expansion [101,102]. Moreover, IL-23 induces the expression of RORγt in Th17 cells, which subsequently promotes the expression of IL-23R and IL-17 [103]. In several experimental psoriasis models, IL-23 acts as a pivotal upstream regulator, bridging innate and adaptive immune responses by promoting the differentiation and maintenance of Th17 cells [104]. When administered to healthy mouse skin, IL-23 triggered the development of psoriasis-like lesions, characterized by epidermal hyperplasia, inflammatory cell infiltration, and the upregulation of several pro-inflammatory cytokines [105,106]. In contrast, IL-23p19−/− mice were protected from imiquimod-induced psoriasis, further emphasizing the critical role of IL-23 [105]. When IL-23 binds to the IL-23R complex, it activates intracellular signaling pathways, including the STAT3 pathway, leading to the production of effector cytokines such as IL-17A, IL-17F, and IL-22. Among these, IL-22 plays a crucial role in promoting keratinocyte hyperproliferation and impairing normal differentiation, resulting in the epidermal thickening (acanthosis) and abnormal cornification (parakeratosis) observed in psoriatic lesions [107]. Furthermore, IL-23 promotes the expansion of other IL-17-producing cells, such as CD8+ T lymphocytes, ILC3s, and γδ T cells, thereby amplifying the inflammatory response [108]. The extreme production of IL-23 in psoriatic skin establishes a self-perpetuating inflammatory loop, in which activated keratinocytes release chemokines that attract additional immune cells, thereby sustaining the inflammatory cascade [109]. In the context of IBD, IL-23 plays a crucial role in promoting the differentiation and expansion of Th17 cells, which are key drivers of chronic intestinal inflammation [110]. In IL-10−/− mice, a preclinical model of IBD, IL-23 has been shown to be vital for the development of chronic intestinal inflammation [111]. IL-23 stimulates the production of pro-inflammatory cytokines such as IL-17A, IL-17F, and IL-22 by Th17 cells and other IL-23R-expressing populations, including γδ T cells and ILCs [112]. These cytokines, in turn, promote neutrophil recruitment, impair epithelial barrier function, and perpetuate the inflammatory cascade [113]. The significance of IL-23 in IBD pathogenesis is supported by genome-wide association studies linking IL23R gene polymorphisms to increased susceptibility to IBD [114,115]. The association has been confirmed in several cohorts which showed a significant association with Crohn’s disease (CD; a subtype of IBD), with the strongest signal for the coding variant Arg381Gln [115]. In a model of ulcerative colitis (UC; another subclass of IBD), the administration of recombinant IL-23 accelerated disease onset and exacerbated severity, highlighting its pro-inflammatory role [116].
Beyond its effects on immune cell dynamics, IL-23 has been implicated in the direct modulation of nociceptive pathways through its influence on peripheral and central sensitization mechanisms [117,118]. Studies have suggested that IL-23 may enhance pain signaling by acting on nociceptive neurons or by indirectly modulating the inflammatory milieu within affected tissues, leading to the highly increased excitability of pain-transmitting pathways [95]. A fundamental element in the pro-nociceptive effects of IL-23 is the action of cyclooxygenases (COX-1 and COX-2). These enzymes play a central role in converting arachidonic acid into bioactive eicosanoids, including prostaglandins (PGs) and thromboxanes, which are established mediators of inflammation and pain [94,95,119]. This interdependence of IL-23 and COX-derived metabolites suggests the existence of multiple converging signaling pathways that synergistically potentiate peripheral and central sensitization mechanisms. The role of COX-generated eicosanoids in nociception is well documented, with prostaglandins (especially PGE2) being key contributors to nociceptor sensitization by reducing their activation threshold and elevating pain perception [120,121].
Studies have indicated that IL-23 plays a key role in the development of arthritis pain, with its activity potentially linked to COX function [93,94,95]. In some experimental models, the injection of IL-23 into the plantar region induced inflammatory pain, which was alleviated by indomethacin [94]. This suggests that IL-23 may contribute to pain development in RA through pathways involving COX-mediated prostaglandin synthesis. On the other hand, although a direct relationship between IL-23 and cyclooxygenases has not yet been established, a notable increase in COX-2 expression has been observed in patients with MS, especially in regions of recent demyelination [122]. In the context of IBD, the interaction between IL-23 and COX may exacerbate inflammation and contribute to pain symptoms. Recent studies have shown that novel 1-H phenyl benzimidazole derivatives, which target the IL-23-mediated inflammatory pathway, can reduce COX-2 activity [123]. With respect to psoriasis, there is no connection between IL-23 and COX-2; however, a recent study marked the relevance of COX-2 in the activity of macrophages [124].
IL-23 has also been implicated in the activation of the lipoxygenase (LOX) pathway, another major branch of arachidonic acid metabolism, which leads to the biosynthesis of leukotrienes [125]. Among these lipid mediators, leukotriene B4 (LTB4) has garnered particular attention due to its ability to activate and recruit innate immune cells, such as neutrophils and macrophages, to sites of inflammation [126,127]. The role of LTB4 in pain extends beyond its pro-inflammatory effects, as it has been shown to directly modulate pain signaling through its interactions with nociceptors. Specifically, LTB4 acts on leukotriene receptors (BLT1 and BLT2), which are expressed on nociceptors and immune cells, leading to the activation of several intracellular signaling cascades that worsen pain hypersensitivity [128,129]. This mechanism is important in chronic inflammatory conditions, such as RA, where IL-23-mediated immune activation contributes to joint destruction and persistent pain [130]. Research has elucidated that IL-23 enhances the production of LTB4 by stimulating the activity of innate immune cells within the synovial microenvironment, thus evoking a self-reinforcing cycle of inflammation [130]. The LOX pathway, especially 5-lipoxygenase (5-LO), plays a role in neuroinflammation and axonal injury in MS [131]. The increased expression of 5-LO has been observed in MS lesions, and elevated leukotriene levels have been detected in the cerebrospinal fluid of MS patients [132]. The inhibition of 5-LO has significantly reduced microglial activation, decreased IL-6 levels, and alleviated axonal injury and motor impairments in several animal studies [133,134,135]. Regarding IL-23, dendritic cells derived from MS patients release higher quantities of IL-23 and exhibit elevated levels of IL-23p19 mRNA, resulting in increased IL-17 production by Th17 cells [136]. On the other hand, some studies have demonstrated the elevated expression of 5-LO pathway proteins in colonic biopsies from patients with IBD, particularly ulcerative colitis (UC). This upregulation correlates with the increased infiltration of some inflammatory cells, such as neutrophils, macrophages, and T cells [137,138,139]. Finally, 12R-lipoxygenase (12R-LOX) contributes to the development of psoriatic lesions. Studies have shown that 12R-LOX is pathologically overexpressed in psoriasis, leading to the strong accumulation of 12R-hydroxyeicosatetraenoic acid (12R-HETE) in the skin [140,141]. The presence of 12R-LOX has been implicated in the proliferative properties of psoriatic skin disorders [142].
Monocytes/macrophages/microglia (Figure 4) are crucial cellular mediators of IL-23-induced nociceptive effects, serving as crucial cellular intermediaries in the inflammatory pain cascade. Experimental strategies involving monocyte and macrophage depletion have provided strong evidence supporting the role of these immune cells in IL-23-induced mechanical hypersensitivity [143,144,145,146]. Some research employing clodronate liposomes (used in a well-established method for the depletion of phagocytic monocytes and macrophages) has demonstrated that the absence of these immune cells effectively prevents IL-23-induced mechanical allodynia and hyperalgesia [147]. These findings highlight that the activity of macrophages is indispensable for the transmission and amplification of IL-23-mediated pain signals. Upon exposure to IL-23, macrophages shift to a pro-inflammatory state, leading to the release of IL-17, a pro-inflammatory cytokine with well-documented effects on immune regulation and pain modulation [148]. IL-17, rather than IL-23 itself, is responsible for the activation of nociceptors [149]. This process occurs through the binding of IL-17 to its cognate receptor, IL-17RA, which is expressed on peripheral sensory neurons, including those within the DRG [150,151]. The strong activation of these nociceptive neurons by IL-17 triggers intracellular signaling cascades, including the activation of MAPKs, NF-κB, and TRPs, such as TRPV1 and TRPA1 [144,152]. Furthermore, the macrophage-derived IL-17 signaling pathway not only contributes to acute nociceptive hypersensitivity but also plays a crucial role in the maintenance and persistence of chronic inflammatory pain states. IL-17 is known to enhance the production of additional pro-inflammatory mediators, including TNF-α and prostaglandins (such as PGE2), all of which sensitize nociceptors and exacerbate pain signaling [153,154,155]. This reinforces the concept that IL-23-induced pain is not solely a transient inflammatory event but rather part of a broader neuroimmune interaction that sustains long-term pain sensitization.
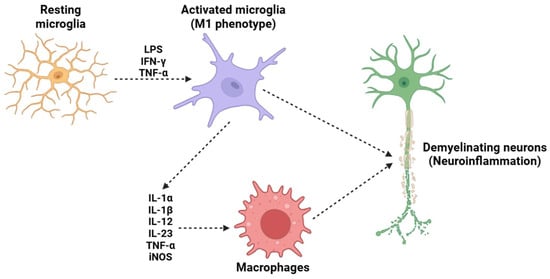
Figure 4.
A potential mechanism underlying the demyelination process in MS is the activation of microglia. Under resting conditions, microglia maintain a resting state; however, in the presence of LPS, IFN-γ, and TNF-α, they adopt a pro-inflammatory M1 phenotype. This phenotypic change induces the release of several pro-inflammatory cytokines (such as IL-1α, IL-1β, IL-12, IL-23, and TNF-α) and the activation of iNOS. This subsequently attracts circulating macrophages, which cross the BBB and migrate into the CNS. The combined effects of resident microglia and circulating macrophages trigger a neuroinflammatory process that ultimately results in the destruction of the neuronal myelin sheath, a critical mechanism in the pathogenesis of MS. Abbreviations: LPS (lipopolysaccharide), IFN-γ (interferon gamma), TNF-α (tumor necrosis factor alpha), IL-1α (interleukin 1 alpha), IL-1β (interleukin 1 beta), IL-12 (interleukin 12), IL-23 (iterleukin 23), and iNOS (inducible nitric oxide synthase).
In conclusion, the interplay between IL-23, macrophages, and IL-17 represents a crucial nexus in the development of inflammatory pain. The indirect activation of nociceptors via macrophage-derived IL-17 highlights the broader concept of neuroimmune crosstalk in pain pathophysiology. Targeting this axis offers promising therapeutic avenues for mitigating inflammatory pain conditions. Further research into these pathways will be crucial for refining targeted interventions and improving the clinical management of chronic inflammatory pain syndromes.
4. Therapeutic Targeting of IL-23 in Inflammatory Diseases
Due to its essential role, therapeutic strategies have been developed to directly target IL-23, aiming to interrupt its signaling pathway. Immunotherapy is a treatment approach focused on boosting the immune system’s ability to fight various diseases, including cancer [156], neuropathies [157], and autoimmune disorders [158]. This approach has gained as a promising alternative to traditional therapies, offering the benefit of fewer side effects [159]. Monoclonal antibody-based immunotherapies have proven effective in managing the progression of numerous chronic conditions by modulating the interactions between immune cells and the PNS/CNS, leading to substantial relief from the painful symptoms associated with these diseases [160].
Table 1 summarizes the most relevant immunotherapeutic treatments targeting IL-23, emphasizing its effectiveness in preventing the onset of various inflammatory diseases and mitigating the severity of pain associated with these conditions.

Table 1.
A list of anti-IL-23 monoclonal antibodies used in clinical trials. Abbreviations: PASI75 (psoriasis area and severity index—75% reduction), ACR20 (American College of Rheumatology—20% improvement), BASDAI20 (bath ankylosing spondylitis disease activity index—20% improvement), BASDAI50 (bath ankylosing spondylitis disease activity index—50% improvement), BASDAI70 (bath ankylosing spondylitis disease activity index—70% improvement), PASI90 (psoriasis area and severity index—90% reduction), PASI100 (psoriasis area and severity index—100% reduction), SF (stool frequency), RB (rectal bleeding), BU (bowel urgency), AP (abdominal pain), and CDAI (Crohn’s disease activity index).
5. Conclusions
This review offers an analysis of the involvement of the IL-23 cytokine in the intricate mechanisms underlying inflammatory pain. As a pro-inflammatory cytokine, IL-23 influences immune cell regulation and the activation of some signaling pathways, contributing to pain sensitization. IL-23 not only intensifies the severity of inflammatory pain but also contributes to its persistence and chronicity, highlighting its potential as a therapeutic target for interventions aimed at mitigating chronic pain and enhancing patient outcomes.
The growing body of evidence supports the notion that modulating IL-23 might offer a promising therapeutic strategy for relieving inflammatory pain. Several studies suggest that therapies targeting IL-23 show potential in relieving pain symptoms. Given the complexity of inflammatory pain and the multifaceted involvement of IL-23 in its pathophysiology, further research is necessary to fully elucidate the mechanisms underlying its biological effects. Additional studies are required to confirm these early findings and assess the long-term efficacy, safety, and side effects of monoclonal anti-IL-23 therapies. Focusing on IL-23R rather than IL-23 itself offers a promising therapeutic approach. Orally bioavailable peptides that specifically target IL-23R may provide superior tissue penetration compared to antibodies, potentially enhancing treatment outcomes for inflammatory diseases [180,181].
In conclusion, this article aims to offer valuable insights to guide the development of innovative and more effective treatment strategies for inflammatory pain. Such advancements could ultimately enhance patient care and significantly improve the quality of life for individuals suffering from chronic inflammatory pain.
Funding
This research received no external funding.
Data Availability Statement
Not applicable. No new data were generated.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 12R-LOX | 12R-lipoxygenase |
| 12R-HETE | 12R-hydroxyeicosatetraenoic acid |
| 5-LO | 5-lipoxygenase |
| ACR20 | American College of Rheumatology (20% improvement) |
| AP | Abdominal pain |
| APC | Antigen-presenting cell |
| BASDAI20 | Bath ankylosing spondylitis disease activity index (20% improvement) |
| BASDAI50 | Bath ankylosing spondylitis disease activity index (50% improvement) |
| BASDAI70 | Bath ankylosing spondylitis disease activity index (70% improvement) |
| BBB | Blood–brain barrier |
| BLT1 | Leukotriene B4 receptor 1 |
| BLT2 | Leukotriene B4 receptor 2 |
| BU | Bowel urgency |
| CCL20 | C-C motif chemokine ligand 20 |
| CCR6 | C-C chemokine receptor type 6 |
| CD | Crohn’s disease |
| CD4 | Cluster of differentiation 4 |
| CD8 | Cluster of differentiation 8 |
| CDAI | Crohn’s disease activity index |
| CNS | Central nervous system |
| COX-1 | Cyclooxygenase 1 |
| COX-2 | Cyclooxygenase 2 |
| CTL | Cytotoxic T cells |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCL9 | C-X-C motif chemokine ligand 9 |
| DAMP | Damage-associated molecular pattern |
| DCs | Dendritic cells |
| dDCs | Dermal dendritic cells |
| DRG | Dorsal root ganglion |
| GALT | Gut-associated lymphoid tissue |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| IBS | Inflammatory bowel syndrome |
| IFN-γ | Interferon gamma |
| IKK | IκB kinase |
| IL-10 | Interleukin 10 |
| IL-12 | Interleukin 12 |
| IL-12p40 | Interleukin 12 p40 subunit |
| IL-12Rβ1 | Interleukin 12 receptor β1 |
| IL-17 | Interleukin 17 |
| IL-17A | Interleukin 17A |
| IL-17F | Interleukin 17F |
| IL-1α | Interleukin 1 alpha |
| IL-1β | Interleukin 1 beta |
| IL-23 | Interleukin 23 |
| IL-23p19 | Interleukin 23 p19 subunit |
| IL-23R | Interleukin 23 receptor |
| IL-27 | Interleukin 27 |
| IL-35 | Interleukin 35 |
| IL-6 | Interleukin 6 |
| ILC | Innate lymphoid cell |
| ILC3 | Innate lymphoid cell 3 |
| iNOS | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| JAK2 | Janus kinase 2 |
| JAK-STAT | Janus kinase-signal transducer and activator of transcription pathway |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| LTB4 | Leukotriene B4 |
| MAIT | Mucosal-associated invariant T cell |
| MAPK | Mitogen-activated protein kinase |
| MDSCs | Myeloid-derived suppressor cells |
| MS | Multiple sclerosis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cell |
| NKT | Natural killer T cell |
| NLR | NOD-like receptor |
| NOD | Nucleotide-binding oligomerization domain |
| p19 | p19 subunit |
| p40 | p40 subunit |
| p50 | p50 subunit |
| p65 | p65 subunit |
| PAMP | Pathogen-associated molecular pattern |
| PASI75 | Psoriasis area and severity index (75% reduction) |
| PASI90 | Psoriasis area and severity index (90% reduction) |
| PASI100 | Psoriasis area and severity index (100% reduction) |
| PG | Prostaglandin |
| PGE2 | Prostaglandin E2 |
| PNS | Peripheral nervous system |
| PRR | Pattern recognition receptor |
| RA | Rheumatoid arthritis |
| RB | Rectal bleeding |
| RORγt | Retinoic acid receptor-related orphan receptor gamma t |
| SF | Stool frequency |
| SPM | Specialized pro-resolving mediator |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-β | Transforming growth factor beta |
| Th | T helper cell |
| Th1 | T helper 1 cell |
| Th17 | T helper 17 cell |
| Th2 | T helper 2 cell |
| TLR | Toll-like receptor |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| TRP | Transient receptor potential |
| TRPA1 | Transient receptor potential ankyrin 1 |
| TRPV1 | Transient receptor potential vanilloid 1 |
| TYK2 | Tyrosine kinase 2 |
| UC | Ulcerative colitis |
| VEGF | Vascular endothelial growth factor |
| γδ T | Gamma delta T cell |
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar]
- Medzhitov, R. The spectrum of inflammatory responses. Science 2021, 374, 1070–1075. [Google Scholar] [PubMed]
- Kiss, A.L. Inflammation in Focus: The Beginning and the End. Pathol. Oncol. Res. 2022, 27, 1610136. [Google Scholar]
- Soares, C.L.R.; Wilairatana, P.; Silva, L.R.; Moreira, P.S.; Vilar Barbosa, N.M.M.; da Silva, P.R.; Coutinho, H.D.M.; de Menezes, I.R.A.; Felipe, C.F.B. Biochemical aspects of the inflammatory process: A narrative review. Biomed. Pharmacother. 2023, 168, 115764. [Google Scholar]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Chávez-Castillo, M.; Ortega, Á.; Cudris-Torres, L.; Duran, P.; Rojas, M.; Manzano, A.; Garrido, B.; Salazar, J.; Silva, A.; Rojas-Gomez, D.M.; et al. Specialized Pro-Resolving Lipid Mediators: The Future of Chronic Pain Therapy? Int. J. Mol. Sci. 2021, 22, 10370. [Google Scholar] [CrossRef]
- Julliard, W.A.; Myo, Y.P.A.; Perelas, A.; Jackson, P.D.; Thatcher, T.H.; Sime, P.J. Specialized pro-resolving mediators as modulators of immune responses. Semin. Immunol. 2022, 59, 101605. [Google Scholar]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Tavakoli, P.; Vollmer-Conna, U.; Hadzi-Pavlovic, D.; Grimm, M.C. A Review of Inflammatory Bowel Disease: A Model of Microbial, Immune and Neuropsychological Integration. Public Health Rev. 2021, 42, 1603990. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Y.; Liu, X. Rheumatoid arthritis: Pathogenesis and therapeutic advances. MedComm 2024, 5, e509. [Google Scholar] [CrossRef]
- Haki, M.; Al-Biati, H.A.; Al-Tameemi, Z.S.; Ali, I.S.; Al-Hussaniy, H.A. Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Medicine 2024, 103, e37297. [Google Scholar] [CrossRef]
- Szewczak, L.; Donskow-Łysoniewska, K. Cytokines and Transgenic Matrix in Autoimmune Diseases: Similarities and Differences. Biomedicines 2020, 8, 559. [Google Scholar] [CrossRef]
- Yasmeen, F.; Pirzada, R.H.; Ahmad, B.; Choi, B.; Choi, S. Understanding Autoimmunity: Mechanisms, Predisposing Factors, and Cytokine Therapies. Int. J. Mol. Sci. 2024, 25, 7666. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, K.D.; Choubey, D. Cytokines in autoimmunity: Role in induction, regulation, and treatment. J. Interferon Cytokine Res. 2011, 31, 695–703. [Google Scholar]
- Abdo, A.I.K.; Tye, G.J. Interleukin 23 and autoimmune diseases: Current and possible future therapies. Inflamm. Res. 2020, 69, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Eldirany, S.A.; Damiani, G.; Ho, M.; Bunick, C.G. Structural Basis for p19 Targeting by Anti-IL-23 Biologics: Correlations with Short- and Long-Term Efficacy in Psoriasis. JID Innov. 2024, 4, 100261. [Google Scholar] [CrossRef]
- Floss, D.M.; Moll, J.M.; Scheller, J. IL-12 and IL-23-Close Relatives with Structural Homologies but Distinct Immunological Functions. Cells 2020, 9, 2184. [Google Scholar] [CrossRef]
- Li, Y.; Yu, X.; Ma, Y.; Hua, S. IL-23 and dendritic cells: What are the roles of their mutual attachment in immune response and immunotherapy? Cytokine 2019, 120, 78–84. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Dong, C. Defining the TH17 cell lineage. Nat. Rev. Immunol. 2021, 21, 618. [Google Scholar]
- Agalioti, T.; Cortesi, F.; Gagliani, N. TH17 cell immune adaptation. Curr. Opin. Immunol. 2023, 83, 102333. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Eyerich, K.; Kuchroo, V.K.; Ritchlin, C.T.; Abreu, M.T.; Elloso, M.M.; Fourie, A.; Fakharzadeh, S.; Sherlock, J.P.; Yang, Y.W.; et al. IL-23 past, present, and future: A roadmap to advancing IL-23 science and therapy. Front. Immunol. 2024, 15, 1331217. [Google Scholar]
- Zhang, W.; Zhong, G.; Ren, X.; Li, M. Research progress of Ustekinumab in the treatment of inflammatory bowel disease. Front. Immunol. 2024, 15, 1322054. [Google Scholar]
- Kerut, C.K.; Wagner, M.J.; Daniel, C.P.; Fisher, C.; Henderson, E.J.; Burroughs, C.R.; Amarasinghe, S.; Willett, O.; Ahmadzadeh, S.; Varrassi, G.; et al. Guselkumab, a Novel Monoclonal Antibody Inhibitor of the p19 Subunit of IL-23, for Psoriatic Arthritis and Plaque Psoriasis: A Review of Its Mechanism, Use, and Clinical Effectiveness. Cureus 2023, 15, e51405. [Google Scholar] [PubMed]
- Banaszczyk, K. Risankizumab in the treatment of psoriasis—Literature review. Reumatologia 2019, 57, 158–162. [Google Scholar]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Hasegawa, H.; Mizoguchi, I.; Chiba, Y.; Ohashi, M.; Xu, M.; Yoshimoto, T. Expanding Diversity in Molecular Structures and Functions of the IL-6/IL-12 Heterodimeric Cytokine Family. Front. Immunol. 2016, 7, 479. [Google Scholar]
- Lupardus, P.J.; Garcia, K.C. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J. Mol. Biol. 2008, 382, 931–941. [Google Scholar]
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity 2018, 48, 45–58.e6. [Google Scholar] [CrossRef] [PubMed]
- Wines, B.D.; Yap, M.L.; Powell, M.S.; Tan, P.S.; Ko, K.K.; Orlowski, E.; Hogarth, P.M. Distinctive expression of interleukin-23 receptor subunits on human Th17 and γδ T cells. Immunol. Cell Biol. 2017, 95, 272–279. [Google Scholar]
- Robinson, R.T. IL12Rβ1: The cytokine receptor that we used to know. Cytokine 2015, 71, 348–359. [Google Scholar]
- Pastor-Fernández, G.; Mariblanca, I.R.; Navarro, M.N. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells 2020, 9, 2044. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. 2018, 9, 1027–1038. [Google Scholar] [PubMed]
- Rachitskaya, A.V.; Hansen, A.M.; Horai, R.; Li, Z.; Villasmil, R.; Luger, D.; Nussenblatt, R.B.; Caspi, R.R. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J. Immunol. 2008, 180, 5167–5171. [Google Scholar]
- Chen, L.; He, Z.; Slinger, E.; Bongers, G.; Lapenda, T.L.S.; Pacer, M.E.; Jiao, J.; Beltrao, M.F.; Soto, A.J.; Harpaz, N.; et al. IL-23 activates innate lymphoid cells to promote neonatal intestinal pathology. Mucosal Immunol. 2015, 8, 390–402. [Google Scholar]
- Wang, H.; Kjer-Nielsen, L.; Shi, M.; D’Souza, C.; Pediongco, T.J.; Cao, H.; Kostenko, L.; Lim, X.Y.; Eckle, S.B.G.; Meehan, B.S.; et al. IL-23 costimulates antigen-specific MAIT cell activation and enables vaccination against bacterial infection. Sci. Immunol. 2019, 4, eaaw0402. [Google Scholar] [CrossRef]
- Glassman, C.R.; Mathiharan, Y.K.; Jude, K.M.; Su, L.; Panova, O.; Lupardus, P.J.; Spangler, J.B.; Ely, L.K.; Thomas, C.; Skiniotis, G.; et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 2021, 184, 983–999.e24. [Google Scholar]
- Che Mat, N.F.; Zhang, X.; Guzzo, C.; Gee, K. Interleukin-23-induced interleukin-23 receptor subunit expression is mediated by the Janus kinase/signal transducer and activation of transcription pathway in human CD4 T cells. J. Interferon Cytokine Res. 2011, 31, 363–371. [Google Scholar]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Lee, P.W.; Smith, A.J.; Yang, Y.; Selhorst, A.J.; Liu, Y.; Racke, M.K.; Lovett-Racke, A.E. IL-23R-activated STAT3/STAT4 is essential for Th1/Th17-mediated CNS autoimmunity. JCI Insight 2017, 2, e91663. [Google Scholar] [PubMed]
- Mortier, C.; Gracey, E.; Coudenys, J.; Manuello, T.; Decruy, T.; Maelegheer, M.; Stappers, F.; Gilis, E.; Gaublomme, D.; Van Hoorebeke, L.; et al. RORγt inhibition ameliorates IL-23 driven experimental psoriatic arthritis by predominantly modulating γδ-T cells. Rheumatology 2023, 62, 3169–3178. [Google Scholar] [PubMed]
- Mitchell, J.P.; Carmody, R.J. NF-κB and the Transcriptional Control of Inflammation. Int. Rev. Cell Mol. Biol. 2018, 335, 41–84. [Google Scholar] [PubMed]
- Liu, D.; Zhong, Z.; Karin, M. NF-κB: A Double-Edged Sword Controlling Inflammation. Biomedicines 2022, 10, 1250. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal. Transduct. Target. Ther. 2024, 9, 53. [Google Scholar]
- Liu, P.; Li, Y.; Wang, W.; Bai, Y.; Jia, H.; Yuan, Z.; Yang, Z. Role and mechanisms of the NF-ĸB signaling pathway in various developmental processes. Biomed. Pharmacother. 2022, 153, 113513. [Google Scholar]
- Barnabei, L.; Laplantine, E.; Mbongo, W.; Rieux-Laucat, F.; Weil, R. NF-κB: At the Borders of Autoimmunity and Inflammation. Front. Immunol. 2021, 12, 716469. [Google Scholar]
- Lindroos, J.; Svensson, L.; Norsgaard, H.; Lovato, P.; Moller, K.; Hagedorn, P.H.; Olsen, G.M.; Labuda, T. IL-23-mediated epidermal hyperplasia is dependent on IL-6. J. Investig. Dermatol. 2011, 131, 1110–1118. [Google Scholar]
- Riol-Blanco, L.; Lazarevic, V.; Awasthi, A.; Mitsdoerffer, M.; Wilson, B.S.; Croxford, A.; Waisman, A.; Kuchroo, V.K.; Glimcher, L.H.; Oukka, M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J. Immunol. 2010, 184, 1710–1720. [Google Scholar]
- Bhattacharya, G.; Sengupta, S.; Jha, R.; Shaw, S.K.; Jogdand, G.M.; Barik, P.K.; Padhan, P.; Parida, J.R.; Devadas, S. IL-21/23 axis modulates inflammatory cytokines and RANKL expression in RA CD4+ T cells via p-Akt1 signaling. Front. Immunol. 2023, 14, 1235514. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Budnick, I.; Singh, M.; Thiruppathi, M.; Alharshawi, K.; Elshabrawy, H.; Holterman, M.J.; Prabhakar, B.S. Dual Role of GM-CSF as a Pro-Inflammatory and a Regulatory Cytokine: Implications for Immune Therapy. J. Interferon Cytokine Res. 2015, 35, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Ten Bergen, L.L.; Petrovic, A.; Krogh Aarebrot, A.; Appel, S. The TNF/IL-23/IL-17 axis-Head-to-head trials comparing different biologics in psoriasis treatment. Scand. J. Immunol. 2020, 92, e12946. [Google Scholar] [CrossRef]
- Korta, A.; Kula, J.; Gomułka, K. The Role of IL-23 in the Pathogenesis and Therapy of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 10172. [Google Scholar] [CrossRef]
- Bao, S.; Zhao, Q.; Zheng, J.; Li, N.; Huang, C.; Chen, M.; Cheng, Q.; Zhu, M.; Yu, K.; Liu, C.; et al. Interleukin-23 mediates the pathogenesis of LPS/GalN-induced liver injury in mice. Int. Immunopharmacol. 2017, 46, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K.; Zirpel, H.; Mruwat, N.; Ludwig, R.J.; Thaci, D. Evaluating the risk of infections under interleukin 23 and interleukin 17 inhibitors relative to tumour necrosis factor inhibitors—A population-based study. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 2319–2326. [Google Scholar] [CrossRef]
- Das, R.; Chen, X.; Komorowski, R.; Hessner, M.J.; Drobyski, W.R. Interleukin-23 secretion by donor antigen-presenting cells is critical for organ-specific pathology in graft-versus-host disease. Blood 2009, 113, 2352–2362. [Google Scholar] [CrossRef]
- Waibler, Z.; Kalinke, U.; Will, J.; Juan, M.H.; Pfeilschifter, J.M.; Radeke, H.H. TLR-ligand stimulated interleukin-23 subunit expression and assembly is regulated differentially in murine plasmacytoid and myeloid dendritic cells. Mol. Immunol. 2007, 44, 1483–1489. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, L.; Chu, Z.; Yang, T.; Sun, H.X.; Yang, F.; Wang, W.; Hou, Y.; Wang, P.; Zhao, Q.; et al. Characterization and biological significance of IL-23-induced neutrophil polarization. Cell. Mol. Immunol. 2018, 15, 518–530. [Google Scholar] [CrossRef]
- Ahmed, A.; Joseph, A.M.; Zhou, J.; Horn, V.; Uddin, J.; Lyu, M.; Goc, J.; JRI Live Cell Bank; Sockolow, R.E.; Wing, J.B.; et al. CTLA-4-expressing ILC3s restrain interleukin-23-mediated inflammation. Nature 2024, 630, 976–983. [Google Scholar]
- Nakamura, A.; Jo, S.; Nakamura, S.; Aparnathi, M.K.; Boroojeni, S.F.; Korshko, M.; Park, Y.S.; Gupta, H.; Vijayan, S.; Rockel, J.S.; et al. HIF-1α and MIF enhance neutrophil-driven type 3 immunity and chondrogenesis in a murine spondyloarthritis model. Cell. Mol. Immunol. 2024, 21, 770–786. [Google Scholar] [PubMed]
- Brailey, P.M.; Evans, L.; López-Rodríguez, J.C.; Sinadinos, A.; Tyrrel, V.; Kelly, G.; O’Donnell, V.; Ghazal, P.; John, S.; Barral, P. CD1d-dependent rewiring of lipid metabolism in macrophages regulates innate immune responses. Nat. Commun. 2022, 13, 6723. [Google Scholar] [PubMed]
- Li, H.; Yao, Q.; Mariscal, A.G.; Wu, X.; Hülse, J.; Pedersen, E.; Helin, K.; Waisman, A.; Vinkel, C.; Thomsen, S.F.; et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat. Commun. 2018, 9, 1420. [Google Scholar] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef]
- Yoshiki, R.; Kabashima, K.; Honda, T.; Nakamizo, S.; Sawada, Y.; Sugita, K.; Yoshioka, H.; Ohmori, S.; Malissen, B.; Tokura, Y.; et al. IL-23 from Langerhans cells is required for the development of imiquimod-induced psoriasis-like dermatitis by induction of IL-17A-producing γδ T cells. J. Investig. Dermatol. 2014, 134, 1912–1921. [Google Scholar]
- Ciccia, F.; Bombardieri, M.; Principato, A.; Giardina, A.; Tripodo, C.; Porcasi, R.; Peralta, S.; Franco, V.; Giardina, E.; Craxi, A.; et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009, 60, 955–965. [Google Scholar] [CrossRef]
- Wallaeys, C.; Garcia-Gonzalez, N.; Libert, C. Paneth cells as the cornerstones of intestinal and organismal health: A primer. EMBO Mol. Med. 2023, 15, e16427. [Google Scholar]
- Noviello, D.; Mager, R.; Roda, G.; Borroni, R.G.; Fiorino, G.; Vetrano, S. The IL23-IL17 Immune Axis in the Treatment of Ulcerative Colitis: Successes, Defeats, and Ongoing Challenges. Front. Immunol. 2021, 12, 611256. [Google Scholar]
- Iliopoulou, L.; Lianopoulou, E.; Kollias, G. IL-23 exerts dominant pathogenic functions in Crohn’s disease-ileitis. Mucosal Immunol. 2024, 17, 769–776. [Google Scholar]
- Schnell, A.; Littman, D.R.; Kuchroo, V.K. TH17 cell heterogeneity and its role in tissue inflammation. Nat. Immunol. 2023, 24, 19–29. [Google Scholar] [CrossRef]
- Stritesky, G.L.; Yeh, N.; Kaplan, M.H. IL-23 promotes maintenance but not commitment to the Th17 lineage. J. Immunol. 2008, 181, 5948–5955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. The role of transforming growth factor β in T helper 17 differentiation. Immunology 2018, 155, 24–35. [Google Scholar] [CrossRef]
- Pan, Y.; Yang, W.; Tang, B.; Wang, X.; Zhang, Q.; Li, W.; Li, L. The protective and pathogenic role of Th17 cell plasticity and function in the tumor microenvironment. Front. Immunol. 2023, 14, 1192303. [Google Scholar] [CrossRef]
- Nalbant, A. IL-17, IL-21, and IL-22 Cytokines of T Helper 17 Cells in Cancer. J. Interferon Cytokine Res. 2019, 39, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Lai, J.H. New insights into the IL-12 and IL-23: From a molecular basis to clinical application in immune-mediated inflammation and cancers. Biochem. Pharmacol. 2020, 175, 113928. [Google Scholar] [CrossRef]
- Subhadarshani, S.; Yusuf, N.; Elmets, C.A. IL-23 and the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1290, 89–98. [Google Scholar] [PubMed]
- Lu, L.; Pan, K.; Zheng, H.X.; Li, J.J.; Qiu, H.J.; Zhao, J.J.; Weng, D.S.; Pan, Q.Z.; Wang, D.D.; Jiang, S.S.; et al. IL-17A promotes immune cell recruitment in human esophageal cancers and the infiltrating dendritic cells represent a positive prognostic marker for patient survival. J. Immunother. 2013, 36, 451–458. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Ngo, V.L.; Geem, D.; Harusato, A.; Hirota, S.A.; Parkos, C.A.; Lukacs, N.W.; Nusrat, A.; Gaboriau-Routhiau, V.; Cerf-Bensussan, N.; et al. IL-17A-mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. 2017, 10, 673–684. [Google Scholar] [CrossRef]
- Hu, J.; Yuan, X.; Belladonna, M.L.; Ong, J.M.; Wachsmann-Hogiu, S.; Farkas, D.L.; Black, K.L.; Yu, J.S. Induction of potent antitumor immunity by intratumoral injection of interleukin 23-transduced dendritic cells. Cancer Res. 2006, 66, 8887–8896. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.; Ye, J.; Ge, Y.; Wang, H.; Dai, E.; Ren, J.; Liu, W.; Ma, C.; Ju, S.; et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics 2021, 11, 6668–6681. [Google Scholar] [CrossRef]
- Yan, J.; Smyth, M.J.; Teng, M.W.L. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a028530. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, X.; Herjan, T.; Li, X. The role of interleukin-17 in tumor development and progression. J. Exp. Med. 2020, 217, e20190297. [Google Scholar] [CrossRef]
- Dadaglio, G.; Fayolle, C.; Oberkampf, M.; Tang, A.; Rudilla, F.; Couillin, I.; Torheim, E.A.; Rosenbaum, P.; Leclerc, C. IL-17 suppresses the therapeutic activity of cancer vaccines through the inhibition of CD8+ T-cell responses. Oncoimmunology 2020, 9, 1758606. [Google Scholar] [CrossRef]
- Yang, B.; Kang, H.; Fung, A.; Zhao, H.; Wang, T.; Ma, D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflamm. 2014, 2014, 623759. [Google Scholar] [CrossRef] [PubMed]
- Numasaki, M.; Fukushi, J.; Ono, M.; Narula, S.K.; Zavodny, P.J.; Kudo, T.; Robbins, P.D.; Tahara, H.; Lotze, M.T. Interleukin-17 promotes angiogenesis and tumor growth. Blood 2003, 101, 2620–2627. [Google Scholar] [CrossRef]
- Huang, Q.; Duan, L.; Qian, X.; Fan, J.; Lv, Z.; Zhang, X.; Han, J.; Wu, F.; Guo, M.; Hu, G.; et al. IL-17 Promotes Angiogenic Factors IL-6, IL-8, and Vegf Production via Stat1 in Lung Adenocarcinoma. Sci. Rep. 2016, 6, 36551. [Google Scholar] [CrossRef] [PubMed]
- Wertheimer, T.; Zwicky, P.; Rindlisbacher, L.; Sparano, C.; Vermeer, M.; de Melo, B.M.S.; Haftmann, C.; Rückert, T.; Sethi, A.; Schärli, S.; et al. IL-23 stabilizes an effector Treg cell program in the tumor microenvironment. Nat. Immunol. 2024, 25, 512–524. [Google Scholar] [CrossRef]
- Qian, X.; Gu, L.; Ning, H.; Zhang, Y.; Hsueh, E.C.; Fu, M.; Hu, X.; Wei, L.; Hoft, D.F.; Liu, J. Increased Th17 cells in the tumor microenvironment is mediated by IL-23 via tumor-secreted prostaglandin E2. J. Immunol. 2013, 190, 5894–5902. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Dikiy, S.; Rudensky, A.Y. Principles of regulatory T cell function. Immunity 2023, 56, 240–255. [Google Scholar] [PubMed]
- Qu, N.; Xu, M.; Mizoguchi, I.; Furusawa, J.; Kaneko, K.; Watanabe, K.; Mizuguchi, J.; Itoh, M.; Kawakami, Y.; Yoshimoto, T. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin. Dev. Immunol. 2013, 2013, 968549. [Google Scholar]
- Lee, K.M.; Lupancu, T.; Chang, L.; Manthey, C.L.; Zeeman, M.; Fourie, A.M.; Hamilton, J.A. The mode of action of IL-23 in experimental inflammatory arthritic pain and disease. Arthritis Res. Ther. 2024, 26, 148. [Google Scholar] [PubMed]
- Lee, K.M.; Zhang, Z.; Achuthan, A.; Fleetwood, A.J.; Smith, J.E.; Hamilton, J.A.; Cook, A.D. IL-23 in arthritic and inflammatory pain development in mice. Arthritis Res. Ther. 2020, 22, 123. [Google Scholar] [PubMed]
- Lee, K.M.; Sherlock, J.P.; Hamilton, J.A. The role of interleukin (IL)-23 in regulating pain in arthritis. Arthritis Res. Ther. 2022, 24, 89. [Google Scholar]
- Lee, K.M.; Lupancu, T.; Achuthan, A.A.; de Steiger, R.; Hamilton, J.A. IL-23p19 in osteoarthritic pain and disease. Osteoarthr. Cartil. 2024, 32, 1413–1418. [Google Scholar]
- Najm, A.; McInnes, I.B. IL-23 orchestrating immune cell activation in arthritis. Rheumatology 2021, 60, iv4–iv15. [Google Scholar] [CrossRef]
- Keystone, E.C.; Schorlemmer, H.U.; Pope, C.; Allison, A.C. Zymosan-induced arthritis: A model of chronic proliferative arthritis following activation of the alternative pathway of complement. Arthritis Rheum. 1977, 20, 1396–1401. [Google Scholar]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar]
- Lee, P.W.; Yang, Y.; Racke, M.K.; Lovett-Racke, A.E. Analysis of TGF-β1 and TGF-β3 as regulators of encephalitogenic Th17 cells: Implications for multiple sclerosis. Brain Behav. Immun. 2015, 46, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Hiltensperger, M.; Korn, T. The Interleukin (IL)-23/T helper (Th)17 Axis in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029637. [Google Scholar] [CrossRef]
- Chi, X.; Jin, W.; Zhao, X.; Xie, T.; Shao, J.; Bai, X.; Jiang, Y.; Wang, X.; Dong, C. RORγt expression in mature TH17 cells safeguards their lineage specification by inhibiting conversion to TH2 cells. Sci. Adv. 2022, 8, eabn7774. [Google Scholar] [CrossRef] [PubMed]
- Whitley, S.K.; Li, M.; Kashem, S.W.; Hirai, T.; Igyártó, B.Z.; Knizner, K.; Ho, J.; Ferris, L.K.; Weaver, C.T.; Cua, D.J.; et al. Local IL-23 is required for proliferation and retention of skin-resident memory TH17 cells. Sci. Immunol. 2022, 7, eabq3254. [Google Scholar] [CrossRef]
- van der Fits, L.; Mourits, S.; Voerman, J.S.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanda, T.; Takaishi, M.; Shiga, T.; Miyoshi, K.; Nakajima, H.; Kamijima, R.; Tarutani, M.; Benson, J.M.; Elloso, M.M.; et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J. Immunol. 2011, 186, 4481–4489. [Google Scholar] [CrossRef]
- Ma, H.L.; Liang, S.; Li, J.; Napierata, L.; Brown, T.; Benoit, S.; Senices, M.; Gill, D.; Dunussi-Joannopoulos, K.; Collins, M.; et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Investig. 2008, 118, 597–607. [Google Scholar] [CrossRef]
- Menter, A.; Krueger, G.G.; Paek, S.Y.; Kivelevitch, D.; Adamopoulos, I.E.; Langley, R.G. Interleukin-17 and Interleukin-23: A Narrative Review of Mechanisms of Action in Psoriasis and Associated Comorbidities. Dermatol. Ther. 2021, 11, 385–400. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Yan, B.Y.; Chan, T.C.; Krueger, J.G. Discovery of the IL-23/IL-17 Signaling Pathway and the Treatment of Psoriasis. J. Immunol. 2018, 201, 1605–1613. [Google Scholar] [CrossRef]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef]
- Liu, Z.; Feng, B.S.; Yang, S.B.; Chen, X.; Su, J.; Yang, P.C. Interleukin (IL)-23 suppresses IL-10 in inflammatory bowel disease. J. Biol. Chem. 2012, 287, 3591–3597. [Google Scholar] [PubMed]
- Ohara, D.; Takeuchi, Y.; Hirota, K. Type 17 immunity: Novel insights into intestinal homeostasis and autoimmune pathogenesis driven by gut-primed T cells. Cell. Mol. Immunol. 2024, 21, 1183–1200. [Google Scholar]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar]
- Xu, W.D.; Xie, Q.B.; Zhao, Y.; Liu, Y. Association of Interleukin-23 receptor gene polymorphisms with susceptibility to Crohn’s disease: A meta-analysis. Sci. Rep. 2015, 5, 18584. [Google Scholar] [CrossRef]
- Tremelling, M.; Cummings, F.; Fisher, S.A.; Mansfield, J.; Gwilliam, R.; Keniry, A.; Nimmo, E.R.; Drummond, H.; Onnie, C.M.; Prescott, N.J.; et al. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology 2007, 132, 1657–1664. [Google Scholar]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [PubMed]
- Ji, J.; He, Q.; Luo, X.; Bang, S.; Matsuoka, Y.; McGinnis, A.; Nackley, A.G.; Ji, R.R. IL-23 Enhances C-Fiber-Mediated and Blue Light-Induced Spontaneous Pain in Female Mice. Front. Immunol. 2021, 12, 787565. [Google Scholar]
- Bian, C.; Wang, Z.C.; Yang, J.L.; Lu, N.; Zhao, Z.Q.; Zhang, Y.Q. Up-regulation of interleukin-23 induces persistent allodynia via CX3CL1 and interleukin-18 signaling in the rat spinal cord after tetanic sciatic stimulation. Brain Behav. Immun. 2014, 37, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar]
- Khanapure, S.P.; Garvey, D.S.; Janero, D.R.; Letts, L.G. Eicosanoids in inflammation: Biosynthesis, pharmacology, and therapeutic frontiers. Curr. Top. Med. Chem. 2007, 7, 311–340. [Google Scholar]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J. Neuroinflamm. 2020, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Alderbi, R.M.; Alam, M.Z.; Alghamdi, B.S.; Alsufiani, H.M.; Abd El-Aziz, G.S.; Omar, U.M.; Al-Ghamdi, M.A. Neurotherapeutic impact of vanillic acid and ibudilast on the cuprizone model of multiple sclerosis. Front. Mol. Neurosci. 2025, 17, 1503396. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, V.; Krishnendu, P.R.; Zachariah, S.M.; Kanthlal, S.K. Novel 1-H Phenyl Benzimidazole Derivatives for IBD Therapy—An in-vitro and in-silico Approach to Evaluate its Effects on the IL-23 Mediated Inflammatory Pathway. Curr. Comput. Aided Drug Des. 2024, 20, 60–71. [Google Scholar]
- Arasa, J.; Terencio, M.C.; Andrés, R.M.; Marín-Castejón, A.; Valcuende-Cavero, F.; Payá, M.; Montesinos, M.C. Defective Induction of COX-2 Expression by Psoriatic Fibroblasts Promotes Pro-inflammatory Activation of Macrophages. Front. Immunol. 2019, 10, 536. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Oyoshi, M.K.; He, R.; Li, Y.; Mondal, S.; Yoon, J.; Afshar, R.; Chen, M.; Lee, D.M.; Luo, H.R.; Luster, A.D.; et al. Leukotriene B4-driven neutrophil recruitment to the skin is essential for allergic skin inflammation. Immunity 2012, 37, 747–758. [Google Scholar] [CrossRef]
- Le Bel, M.; Brunet, A.; Gosselin, J. Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J. Innate Immun. 2014, 6, 159–168. [Google Scholar]
- Kiyoyuki, Y.; Taniguchi, W.; Okubo, M.; Yamanaka, H.; Kobayashi, K.; Nishio, N.; Nakatsuka, T.; Noguchi, K. Leukotriene enhances NMDA-induced inward currents in dorsal horn neurons of the rat spinal cord after peripheral nerve injury. Mol. Pain 2015, 11, 53. [Google Scholar] [CrossRef]
- Asahara, M.; Ito, N.; Hoshino, Y.; Sasaki, T.; Yokomizo, T.; Nakamura, M.; Shimizu, T.; Yamada, Y. Role of leukotriene B4 (LTB4)-LTB4 receptor 1 signaling in post-incisional nociceptive sensitization and local inflammation in mice. PLoS ONE 2022, 17, e0276135. [Google Scholar] [CrossRef]
- Zheng, L.X.; Li, K.X.; Hong, F.F.; Yang, S.L. Pain and bone damage in rheumatoid arthritis: Role of leukotriene B4. Clin. Exp. Rheumatol. 2019, 37, 872–878. [Google Scholar]
- Broos, J.Y.; van der Burgt, R.T.M.; Konings, J.; Rijnsburger, M.; Werz, O.; de Vries, H.E.; Giera, M.; Kooij, G. Arachidonic acid-derived lipid mediators in multiple sclerosis pathogenesis: Fueling or dampening disease progression? J. Neuroinflamm. 2024, 21, 21. [Google Scholar]
- Pietrantonio, F.; Serreqi, A.; Zerbe, H.; Svenningsson, P.; Aigner, L. The leukotriene receptor antagonist montelukast as a potential therapeutic adjuvant in multiple sclerosis—A review. Front. Pharmacol. 2024, 15, 1450493. [Google Scholar]
- Emerson, M.R.; LeVine, S.M. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res. 2004, 1021, 140–145. [Google Scholar] [PubMed]
- Yoshikawa, K.; Palumbo, S.; Toscano, C.D.; Bosetti, F. Inhibition of 5-lipoxygenase activity in mice during cuprizone-induced demyelination attenuates neuroinflammation, motor dysfunction and axonal damage. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 43–52. [Google Scholar]
- Han, B.; Zhang, Y.Y.; Ye, Z.Q.; Xiao, Y.; Rasouli, J.; Wu, W.C.; Ye, S.M.; Guo, X.Y.; Zhu, L.; Rostami, A.; et al. Montelukast alleviates inflammation in experimental autoimmune encephalomyelitis by altering Th17 differentiation in a mouse model. Immunology 2021, 163, 185–200. [Google Scholar]
- Li, Y.; Chu, N.; Hu, A.; Gran, B.; Rostami, A.; Zhang, G.X. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain 2007, 130, 490–501. [Google Scholar]
- Jupp, J.; Hillier, K.; Elliott, D.H.; Fine, D.R.; Bateman, A.C.; Johnson, P.A.; Cazaly, A.M.; Penrose, J.F.; Sampson, A.P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 537–546. [Google Scholar] [PubMed]
- Starr, A.E.; Deeke, S.A.; Ning, Z.; Chiang, C.K.; Zhang, X.; Mottawea, W.; Singleton, R.; Benchimol, E.I.; Wen, M.; Mack, D.R.; et al. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn’s disease from UC. Gut 2017, 66, 1573–1583. [Google Scholar]
- Zhang, C.; Zhang, J.; Zhang, Y.; Song, Z.; Bian, J.; Yi, H.; Ma, Z. Identifying neutrophil-associated subtypes in ulcerative colitis and confirming neutrophils promote colitis-associated colorectal cancer. Front. Immunol. 2023, 14, 1095098. [Google Scholar]
- Boeglin, W.E.; Kim, R.B.; Brash, A.R. A 12R-lipoxygenase in human skin: Mechanistic evidence, molecular cloning, and expression. Proc. Natl. Acad. Sci. USA 1998, 95, 6744–6749. [Google Scholar]
- Fürstenberger, G.; Epp, N.; Eckl, K.M.; Hennies, H.C.; Jørgensen, C.; Hallenborg, P.; Kristiansen, K.; Krieg, P. Role of epidermis-type lipoxygenases for skin barrier function and adipocyte differentiation. Prostaglandins Other Lipid Mediat. 2007, 82, 128–134. [Google Scholar] [PubMed]
- Bhuktar, H.; Shukla, S.; Kakularam, K.R.; Battu, S.; Srikanth, M.; Srivastava, S.; Medishetti, R.; Ram, P.; Jagadish, P.C.; Rasool, M.; et al. Design, synthesis and evaluation of 2-aryl quinoline derivatives against 12R-lipoxygenase (12R-LOX): Discovery of first inhibitor of 12R-LOX. Bioorg. Chem. 2023, 138, 106606. [Google Scholar]
- Weaver, C.T.; Hatton, R.D.; Mangan, P.R.; Harrington, L.E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007, 25, 821–852. [Google Scholar]
- Luo, X.; Chen, O.; Wang, Z.; Bang, S.; Ji, J.; Lee, S.H.; Huh, Y.; Furutani, K.; He, Q.; Tao, X.; et al. IL-23/IL-17A/TRPV1 axis produces mechanical pain via macrophage-sensory neuron crosstalk in female mice. Neuron 2021, 109, 2691–2706.e5. [Google Scholar]
- Chen, J.; Liao, M.Y.; Gao, X.L.; Zhong, Q.; Tang, T.T.; Yu, X.; Liao, Y.H.; Cheng, X. IL-17A induces pro-inflammatory cytokines production in macrophages via MAPKinases, NF-κB and AP-1. Cell. Physiol. Biochem. 2013, 32, 1265–1274. [Google Scholar]
- Tan, Z.; Lin, Z.J.; Wu, L.J.; Zhou, L.J. The Macrophage IL-23/IL-17A Pathway: A New Neuro-Immune Mechanism in Female Mechanical Pain. Neurosci. Bull. 2022, 38, 453–455. [Google Scholar]
- Ward, N.L.; Loyd, C.M.; Wolfram, J.A.; Diaconu, D.; Michaels, C.M.; McCormick, T.S. Depletion of antigen-presenting cells by clodronate liposomes reverses the psoriatic skin phenotype in KC-Tie2 mice. Br. J. Dermatol. 2011, 164, 750–758. [Google Scholar]
- Jiang, X.; Zhou, R.; Zhang, Y.; Zhu, T.; Li, Q.; Zhang, W. Interleukin-17 as a potential therapeutic target for chronic pain. Front. Immunol. 2022, 13, 999407. [Google Scholar]
- Richter, F.; Natura, G.; Ebbinghaus, M.; von Banchet, G.S.; Hensellek, S.; König, C.; Bräuer, R.; Schaible, H.G. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum. 2012, 64, 4125–4134. [Google Scholar]
- Schaible, H.G. Nociceptive neurons detect cytokines in arthritis. Arthritis Res. Ther. 2014, 16, 470. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, Y.; Lao, L.; Saito, R.; Li, A.; Bäckman, C.M.; Berman, B.M.; Ren, K.; Wei, P.K.; Zhang, R.X. Spinal interleukin-17 promotes thermal hyperalgesia and NMDA NR1 phosphorylation in an inflammatory pain rat model. Pain 2013, 154, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Wen, C.H.; Wang, M.; Xiao, Z.D.; Zhang, Z.Z.; Wu, C.L.; Wu, R. IL-23/IL-17 immune axis mediates the imiquimod-induced psoriatic inflammation by activating ACT1/TRAF6/TAK1/NF-κB pathway in macrophages and keratinocytes. Kaohsiung J. Med. Sci. 2023, 39, 789–800. [Google Scholar] [CrossRef]
- Polese, B.; Thurairajah, B.; Zhang, H.; Soo, C.L.; McMahon, C.A.; Fontes, G.; Hussain, S.N.A.; Abadie, V.; King, I.L. Prostaglandin E2 amplifies IL-17 production by γδ T cells during barrier inflammation. Cell Rep. 2021, 36, 109456. [Google Scholar] [CrossRef]
- McNamee, K.E.; Alzabin, S.; Hughes, J.P.; Anand, P.; Feldmann, M.; Williams, R.O.; Inglis, J.J. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain 2011, 152, 1838–1845. [Google Scholar] [CrossRef]
- Mu, X.; Gu, R.; Tang, M.; Wu, X.; He, W.; Nie, X. IL-17 in wound repair: Bridging acute and chronic responses. Cell Commun. Signal. 2024, 22, 288. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar]
- Léger, J.M.; Guimarães-Costa, R.; Muntean, C. Immunotherapy in Peripheral Neuropathies. Neurotherapeutics 2016, 13, 96–107. [Google Scholar] [CrossRef]
- Rakshit, S.; Molina, J.R. Immunotherapy in patients with autoimmune disease. J. Thorac. Dis. 2020, 12, 7032–7038. [Google Scholar] [CrossRef]
- Lees, J.G.; Duffy, S.S.; Moalem-Taylor, G. Immunotherapy targeting cytokines in neuropathic pain. Front. Pharmacol. 2013, 4, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Huh, Y.; Bortsov, A.; Diatchenko, L.; Ji, R.R. Immunotherapies in chronic pain through modulation of neuroimmune interactions. Pharmacol. Ther. 2023, 248, 108476. [Google Scholar] [CrossRef]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [PubMed]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B.; PHOENIX 1 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.C.; Wang, Y.; Li, S.; et al. PHOENIX 2 study investigators. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [Google Scholar]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; McInnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.K.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann. Rheum. Dis. 2014, 73, 990–999. [Google Scholar] [PubMed]
- Kimball, A.B.; Gordon, K.B.; Langley, R.G.; Menter, A.; Chartash, E.K.; Valdes, J.; ABT-874 Psoriasis Study Investigators. Safety and efficacy of ABT-874, a fully human interleukin 12/23 monoclonal antibody, in the treatment of moderate to severe chronic plaque psoriasis: Results of a randomized, placebo-controlled, phase 2 trial. Arch. Dermatol. 2008, 144, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, R.; Thirthar Palanivelu, V. Tildrakizumab for the treatment of psoriasis. Expert Rev. Clin. Immunol. 2019, 15, 5–12. [Google Scholar]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.; Randazzo, B.; Wasfi, Y.; Shen, Y.K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar]
- Gordon, K.B.; Strober, B.; Lebwohl, M.; Augustin, M.; Blauvelt, A.; Poulin, Y.; Papp, K.A.; Sofen, H.; Puig, L.; Foley, P.; et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): Results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 2018, 392, 650–661. [Google Scholar]
- Louis, E.; Schreiber, S.; Panaccione, R.; Bossuyt, P.; Biedermann, L.; Colombel, J.F.; Parkes, G.; Peyrin-Biroulet, L.; D’Haens, G.; Hisamatsu, T.; et al. Risankizumab for Ulcerative Colitis: Two Randomized Clinical Trials. JAMA 2024, 332, 881–897. [Google Scholar]
- Peyrin-Biroulet, L.; Allegretti, J.R.; Rubin, D.T.; Bressler, B.; Germinaro, M.; Huang, K.G.; Shipitofsky, N.; Zhang, H.; Wilson, R.; Han, C.; et al. Guselkumab in Patients With Moderately to Severely Active Ulcerative Colitis: QUASAR Phase 2b Induction Study. Gastroenterology 2023, 165, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Dignass, A.; Matsuoka, K.; Ferrante, M.; Long, M.; Redondo, I.; Moses, R.; Maier, S.; Hunter Gibble, T.; Morris, N.; et al. Early and Sustained Symptom Control with Mirikizumab in Patients with Ulcerative Colitis in the Phase 3 LUCENT Programme. J. Crohns. Colitis 2024, 18, 1845–1856. [Google Scholar] [CrossRef]
- Panaccione, R.; Sandborn, W.J.; Gordon, G.L.; Lee, S.D.; Safdi, A.; Sedghi, S.; Feagan, B.G.; Hanauer, S.; Reinisch, W.; Valentine, J.F.; et al. Briakinumab for treatment of Crohn’s disease: Results of a randomized trial. Inflamm. Bowel Dis. 2015, 21, 1329–1340. [Google Scholar] [PubMed]
- D’Haens, G.; Panaccione, R.; Baert, F.; Bossuyt, P.; Colombel, J.F.; Danese, S.; Dubinsky, M.; Feagan, B.G.; Hisamatsu, T.; Lim, A.; et al. Risankizumab as induction therapy for Crohn’s disease: Results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet 2022, 399, 2015–2030. [Google Scholar] [CrossRef]
- Ferrante, M.; Panaccione, R.; Baert, F.; Bossuyt, P.; Colombel, J.F.; Danese, S.; Dubinsky, M.; Feagan, B.G.; Hisamatsu, T.; Lim, A.; et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: Results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet 2022, 399, 2031–2046. [Google Scholar] [CrossRef]
- Sandborn, W.J.; D’Haens, G.R.; Reinisch, W.; Panés, J.; Chan, D.; Gonzalez, S.; Weisel, K.; Germinaro, M.; Frustaci, M.E.; Yang, Z.; et al. Guselkumab for the Treatment of Crohn’s Disease: Induction Results From the Phase 2 GALAXI-1 Study. Gastroenterology 2022, 162, 1650–1664.e8. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Panaccione, R.; Feagan, B.G.; Afzali, A.; Rubin, D.T.; Sands, B.E.; Reinisch, W.; Panés, J.; Sahoo, A.; Terry, N.A.; et al. Efficacy and safety of 48 weeks of guselkumab for patients with Crohn’s disease: Maintenance results from the phase 2, randomised, double-blind GALAXI-1 trial. Lancet Gastroenterol. Hepatol. 2024, 9, 133–146. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Kierkus, J.; Higgins, P.D.R.; Fischer, M.; Jairath, V.; Hirai, F.; D’Haens, G.; Belin, R.M.; Miller, D.; et al. Efficacy and Safety of Mirikizumab in a Randomized Phase 2 Study of Patients With Crohn’s Disease. Gastroenterology 2022, 162, 495–508. [Google Scholar] [CrossRef]
- Ferrante, M.; D’Haens, G.; Jairath, V.; Danese, S.; Chen, M.; Ghosh, S.; Hisamatsu, T.; Kierkus, J.; Siegmund, B.; Bragg, S.M.; et al. Efficacy and safety of mirikizumab in patients with moderately-to-severely active Crohn’s disease: A phase 3, multicentre, randomised, double-blind, placebo-controlled and active-controlled, treat-through study. Lancet 2024, 404, 2423–2436. [Google Scholar] [CrossRef]
- Quiniou, C.; Domínguez-Punaro, M.; Cloutier, F.; Erfani, A.; Ennaciri, J.; Sivanesan, D.; Sanchez, M.; Chognard, G.; Hou, X.; Rivera, J.C.; et al. Specific targeting of the IL-23 receptor, using a novel small peptide noncompetitive antagonist, decreases the inflammatory response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1216–R1230. [Google Scholar] [CrossRef]
- Cheng, X.; Lee, T.; Ledet, G.; Zemade, G.; Tovera, J.; Campbell, R.; Purro, N.; Annamalai, T.; Masjedizadeh, M.; Liu, D.; et al. 751 Safety, Tolerability, and Pharmacokinetics of PTG-200, an Oral GI-Restricted Peptide Antagonist of IL-23 Receptor, in Normal Healthy Volunteers. Am. J. Gastroenterol. 2019, 114, S439–S440. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).