
Compensatory Regulation of Excitation/Inhibition Balance in the Ventral Hippocampus: Insights from Fragile X Syndrome


{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Excitation/Inhibition Balance
2.1. Basic Mechanisms of the E/I Balance
2.2. Homeostatic Regulation of the E/I Balance
2.3. The E/I Balance in Neuropsychiatric and Neurodevelopmental Disorders
3. The E/I Balance in the Disordered Hippocampus
3.1. The E/I Balance in the Epileptic Hippocampus
3.2. The E/I Balance in the FXS Hippocampus
4. Dorsoventral Organization of the Hippocampus
4.1. Functional Specialization Along the Hippocampus
4.2. Dorsoventral Circuit Diversification in the FXS Hippocampus
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koolschijn, R.S.; Shpektor, A.; Clarke, W.T.; Ip, I.B.; Dupret, D.; Emir, U.E.; Barron, H.C. Memory recall involves a transient break in excitatory-inhibitory balance. eLife 2021, 10, e70071. [Google Scholar] [CrossRef] [PubMed]
- Sakimoto, Y.; Oo, P.M.; Goshima, M.; Kanehisa, I.; Tsukada, Y.; Mitsushima, D. Significance of GABA(A) Receptor for Cognitive Function and Hippocampal Pathology. Int. J. Mol. Sci. 2021, 22, 12456. [Google Scholar] [CrossRef]
- Froemke, R.C. Plasticity of cortical excitatory-inhibitory balance. Annu. Rev. Neurosci. 2015, 38, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.R.; Gao, W.J. PV Interneurons: Critical Regulators of E/I Balance for Prefrontal Cortex-Dependent Behavior and Psychiatric Disorders. Front. Neural Circuits 2018, 12, 37. [Google Scholar] [CrossRef]
- Haider, B.; Häusser, M.; Carandini, M. Inhibition dominates sensory responses in the awake cortex. Nature 2013, 493, 97. [Google Scholar] [CrossRef]
- Vogels, T.P.; Sprekeler, H.; Zenke, F.; Clopath, C.; Gerstner, W. Inhibitory plasticity balances excitation and inhibition in sensory pathways and memory networks. Science 2011, 334, 1569–1573. [Google Scholar] [CrossRef]
- van Vreeswijk, C.; Sompolinsky, H. Chaos in neuronal networks with balanced excitatory and inhibitory activity. Science 1996, 274, 1724–1726. [Google Scholar]
- Haider, B.; Duque, A.; Hasenstaub, A.R.; McCormick, D.A. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J. Neurosci. 2006, 26, 4535–4545. [Google Scholar]
- Isaacson, J.S.; Scanziani, M. How inhibition shapes cortical activity. Neuron 2011, 72, 231–243. [Google Scholar] [CrossRef]
- Uliana, D.L.; Lisboa, J.R.F.; Gomes, F.V.; Grace, A.A. The excitatory-inhibitory balance as a target for the development of novel drugs to treat schizophrenia. Biochem. Pharmacol. 2024, 228, 116298. [Google Scholar] [CrossRef]
- Milovanovic, M.; Grujicic, R. Electroencephalography in Assessment of Autism Spectrum Disorders: A Review. Front. Psychiatry 2021, 12, 686021. [Google Scholar] [CrossRef]
- Fenton, A.A. Excitation-inhibition discoordination in rodent models of mental disorders. Biol. Psychiatry 2015, 77, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Bülow, P.; Segal, M.; Bassell, G.J. Mechanisms Driving the Emergence of Neuronal Hyperexcitability in Fragile X Syndrome. Int. J. Mol. Sci. 2022, 23, 6315. [Google Scholar] [CrossRef]
- Liu, X.; Kumar, V.; Tsai, N.P.; Auerbach, B.D. Hyperexcitability and Homeostasis in Fragile X Syndrome. Front. Mol. Neurosci. 2021, 14, 805929. [Google Scholar] [CrossRef]
- Ghatak, S.; Talantova, M.; McKercher, S.R.; Lipton, S.A. Novel Therapeutic Approach for Excitatory/Inhibitory Imbalance in Neurodevelopmental and Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 701–721. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.H.; Chau, C.Q.; Kamel, N.; Thanh, H.H.T.; Yahya, N. Functional excitation-inhibition ratio for social anxiety analysis and severity assessment. Front. Psychiatry 2024, 15, 1461290. [Google Scholar] [CrossRef]
- Scharfman, H.E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 2007, 7, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Eichler, S.A.; Meier, J.C. E-I balance and human diseases—From molecules to networking. Front. Mol. Neurosci. 2008, 1, 195. [Google Scholar] [CrossRef]
- Xing, W.; de Lima, A.D.; Voigt, T. The Structural E/I Balance Constrains the Early Development of Cortical Network Activity. Front. Cell. Neurosci. 2021, 15, 687306. [Google Scholar] [CrossRef]
- Yang, W.; Sun, Q.Q. Circuit-specific and neuronal subcellular-wide E-I balance in cortical pyramidal cells. Sci. Rep. 2018, 8, 3971. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Tjia, M.; Thapliyal, S. Homeostatic plasticity and excitation-inhibition balance: The good, the bad, and the ugly. Curr. Opin. Neurobiol. 2022, 75, 102553. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, H.; Jiang, T.; Yu, S. Natural brain state change with E/I balance shifting toward inhibition is associated with vigilance impairment. iScience 2023, 26, 107963. [Google Scholar] [CrossRef]
- Zhou, S.; Yu, Y. Synaptic E-I Balance Underlies Efficient Neural Coding. Front. Neurosci. 2018, 12, 46. [Google Scholar] [CrossRef]
- Wen, W.; Turrigiano, G.G. Keeping Your Brain in Balance: Homeostatic Regulation of Network Function. Annu. Rev. Neurosci. 2024, 47, 41–61. [Google Scholar] [CrossRef]
- Sukenik, N.; Vinogradov, O.; Weinreb, E.; Segal, M.; Levina, A.; Moses, E. Neuronal circuits overcome imbalance in excitation and inhibition by adjusting connection numbers. Proc. Natl. Acad. Sci. USA 2021, 118, e2018459118. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.T.; Fanselow, M.S. Fear and memory: A view of the hippocampus through the lens of the amygdala. In Space, Time and Memory in the Hippocampal Formation; Springer: Berlin/Heidelberg, Germany, 2014; pp. 465–496. [Google Scholar]
- Gulyaeva, N.V.J.B. Stress-associated molecular and cellular hippocampal mechanisms common for epilepsy and comorbid depressive disorders. Biochemistry 2021, 86, 641–656. [Google Scholar]
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; McHugh, S.B.; Rawlins, J.N.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014, 15, 181–192. [Google Scholar]
- Okuyama, T.; Kitamura, T.; Roy, D.S.; Itohara, S.; Tonegawa, S. Ventral CA1 neurons store social memory. Science 2016, 353, 1536–1541. [Google Scholar] [PubMed]
- Shi, H.J.; Wang, S.; Wang, X.P.; Zhang, R.X.; Zhu, L.J. Hippocampus: Molecular, Cellular, and Circuit Features in Anxiety. Neurosci. Bull. 2023, 39, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Dedovic, K.; Duchesne, A.; Andrews, J.; Engert, V.; Pruessner, J.C. The brain and the stress axis: The neural correlates of cortisol regulation in response to stress. Neuroimage 2009, 47, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef]
- Ruggiero, R.N.; Rossignoli, M.T.; Marques, D.B.; de Sousa, B.M.; Romcy-Pereira, R.N.; Lopes-Aguiar, C.; Leite, J.P. Neuromodulation of Hippocampal-Prefrontal Cortical Synaptic Plasticity and Functional Connectivity: Implications for Neuropsychiatric Disorders. Front. Cell. Neurosci. 2021, 15, 732360. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2018, 44, 1306–1322. [Google Scholar] [CrossRef]
- Sloviter, R.S. Hippocampal pathology and pathophysiology in temporal lobe epilepsy. Neurologia 1996, 11 (Suppl. S4), 29–32. [Google Scholar]
- Li, Y.; Shen, M.; Stockton, M.E.; Zhao, X. Hippocampal deficits in neurodevelopmental disorders. Neurobiol. Learn. Mem. 2019, 165, 106945. [Google Scholar] [CrossRef]
- Bernasconi, N.; Bernasconi, A.; Caramanos, Z.; Antel, S.B.; Andermann, F.; Arnold, D.L. Mesial temporal damage in temporal lobe epilepsy: A volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain 2003, 126, 462–469. [Google Scholar] [CrossRef]
- Spencer, D.D.; Spencer, S.S.; Mattson, R.H.; Williamson, P.D.; Novelly, R.A. Access to the posterior medial temporal lobe structures in the surgical treatment of temporal lobe epilepsy. Neurosurgery 1984, 15, 667–671. [Google Scholar] [CrossRef]
- Babb, T.L.; Brown, W.J.; Pretorius, J.; Davenport, C.; Lieb, J.P.; Crandall, P.H. Temporal lobe volumetric cell densities in temporal lobe epilepsy. Epilepsia 1984, 25, 729–740. [Google Scholar] [CrossRef]
- Quigg, M.; Bertram, E.H.; Jackson, T. Longitudinal distribution of hippocampal atrophy in mesial temporal lobe epilepsy. Epilepsy Res. 1997, 27, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Schobel, S.A.; Kelly, M.A.; Corcoran, C.M.; Van Heertum, K.; Seckinger, R.; Goetz, R.; Harkavy-Friedman, J.; Malaspina, D. Anterior hippocampal and orbitofrontal cortical structural brain abnormalities in association with cognitive deficits in schizophrenia. Schizophr. Res. 2009, 114, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.Y.; Chambers, R.A.; Lipska, B.K. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav. Brain Res. 2009, 204, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Strange, B.A.; Witter, M.P.; Lein, E.S.; Moser, E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014, 15, 655–669. [Google Scholar] [CrossRef]
- Papatheodoropoulos, C. Electrophysiological evidence for long-axis intrinsic diversification of the hippocampus. Front. Biosci. 2018, 23, 109–145. [Google Scholar] [CrossRef]
- Leontiadis, L.J.; Trompoukis, G.; Tsotsokou, G.; Miliou, A.; Felemegkas, P.; Papatheodoropoulos, C. Rescue of sharp wave-ripples and prevention of network hyperexcitability in the ventral but not the dorsal hippocampus of a rat model of fragile X syndrome. Front. Cell. Neurosci. 2023, 17, 1296235. [Google Scholar] [CrossRef]
- Leontiadis, L.J.; Trompoukis, G.; Felemegkas, P.; Tsotsokou, G.; Miliou, A.; Papatheodoropoulos, C. Increased Inhibition May Contribute to Maintaining Normal Network Function in the Ventral Hippocampus of a Fmr1-Targeted Transgenic Rat Model of Fragile X Syndrome. Brain Sci. 2023, 13, 1598. [Google Scholar] [CrossRef]
- Ntoulas, G.; Brakatselos, C.; Nakas, G.; Asprogerakas, M.Z.; Delis, F.; Leontiadis, L.J.; Trompoukis, G.; Papatheodoropoulos, C.; Gkikas, D.; Valakos, D.; et al. Multi-level profiling of the Fmr1 KO rat unveils altered behavioral traits along with aberrant glutamatergic function. Transl. Psychiatry 2024, 14, 104. [Google Scholar] [CrossRef]
- Balkenhol, J.; Händel, B.; Biswas, S.; Grohmann, J.; Kistowski, J.V.; Prada, J.; Bosman, C.A.; Ehrenreich, H.; Wojcik, S.M.; Kounev, S.; et al. Beyond-local neural information processing in neuronal networks. Comput. Struct. Biotechnol. J. 2024, 23, 4288–4305. [Google Scholar] [CrossRef]
- Lerner, T.N.; Ye, L.; Deisseroth, K. Communication in Neural Circuits: Tools, Opportunities, and Challenges. Cell 2016, 164, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Abbott, L.F.; Regehr, W.G. Synaptic computation. Nature 2004, 431, 796–803. [Google Scholar] [CrossRef]
- Silver, R.A. Neuronal arithmetic. Nat. Rev. Neurosci. 2010, 11, 474–489. [Google Scholar] [CrossRef]
- Pérez-Ortega, J.; Alejandre-García, T.; Yuste, R. Long-term stability of cortical ensembles. eLife 2021, 10, e64449. [Google Scholar] [CrossRef]
- Jensen, K.T.; Kadmon Harpaz, N.; Dhawale, A.K.; Wolff, S.B.E.; Ölveczky, B.P. Long-term stability of single neuron activity in the motor system. Nat. Neurosci. 2022, 25, 1664–1674. [Google Scholar] [CrossRef]
- Grangeray-Vilmint, A.; Valera, A.M.; Kumar, A.; Isope, P. Short-Term Plasticity Combines with Excitation-Inhibition Balance to Expand Cerebellar Purkinje Cell Dynamic Range. J. Neurosci. 2018, 38, 5153–5167. [Google Scholar] [CrossRef]
- Bhatia, A.; Moza, S.; Bhalla, U.S. Precise excitation-inhibition balance controls gain and timing in the hippocampus. eLife 2019, 8, e43415. [Google Scholar] [CrossRef] [PubMed]
- Atallah, B.V.; Scanziani, M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron 2009, 62, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Okun, M.; Lampl, I. Instantaneous correlation of excitation and inhibition during ongoing and sensory-evoked activities. Nat. Neurosci. 2008, 11, 535–537. [Google Scholar] [CrossRef]
- Vogels, T.P.; Abbott, L.F. Gating multiple signals through detailed balance of excitation and inhibition in spiking networks. Nat. Neurosci. 2009, 12, 483–491. [Google Scholar] [CrossRef]
- Taub, A.H.; Katz, Y.; Lampl, I. Cortical balance of excitation and inhibition is regulated by the rate of synaptic activity. J. Neurosci. 2013, 33, 14359–14368. [Google Scholar] [CrossRef] [PubMed]
- Foss-Feig, J.H.; Adkinson, B.D.; Ji, J.L.; Yang, G.; Srihari, V.H.; McPartland, J.C.; Krystal, J.H.; Murray, J.D.; Anticevic, A. Searching for Cross-Diagnostic Convergence: Neural Mechanisms Governing Excitation and Inhibition Balance in Schizophrenia and Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S. Keeping Excitation-Inhibition Ratio in Balance. Int. J. Mol. Sci. 2022, 23, 5746. [Google Scholar] [CrossRef]
- Selimbeyoglu, A.; Kim, C.K.; Inoue, M.; Lee, S.Y.; Hong, A.S.O.; Kauvar, I.; Ramakrishnan, C.; Fenno, L.E.; Davidson, T.J.; Wright, M.; et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med. 2017, 9, eaah6733. [Google Scholar] [CrossRef]
- Xue, M.; Atallah, B.V.; Scanziani, M. Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 2014, 511, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.F.; Dobrunz, L.E. Short-term plasticity regulates the excitation/inhibition ratio and the temporal window for spike integration in CA1 pyramidal cells. Eur. J. Neurosci. 2015, 41, 1402–1415. [Google Scholar] [CrossRef]
- Howes, O.D.; Shatalina, E. Integrating the Neurodevelopmental and Dopamine Hypotheses of Schizophrenia and the Role of Cortical Excitation-Inhibition Balance. Biol. Psychiatry 2022, 92, 501–513. [Google Scholar] [CrossRef]
- Tatti, R.; Haley, M.S.; Swanson, O.K.; Tselha, T.; Maffei, A. Neurophysiology and Regulation of the Balance Between Excitation and Inhibition in Neocortical Circuits. Biol. Psychiatry 2017, 81, 821–831. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Nelson, S.B. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004, 5, 97–107. [Google Scholar] [CrossRef]
- Desai, N.S. Homeostatic plasticity in the CNS: Synaptic and intrinsic forms. J. Physiol. Paris 2003, 97, 391–402. [Google Scholar] [CrossRef]
- Peng, Y.R.; Zeng, S.Y.; Song, H.L.; Li, M.Y.; Yamada, M.K.; Yu, X. Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J. Neurosci. 2010, 30, 16220–16231. [Google Scholar] [CrossRef] [PubMed]
- Liu, G. Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat. Neurosci. 2004, 7, 373–379. [Google Scholar] [CrossRef]
- Campanac, E.; Gasselin, C.; Baude, A.; Rama, S.; Ankri, N.; Debanne, D. Enhanced intrinsic excitability in basket cells maintains excitatory-inhibitory balance in hippocampal circuits. Neuron 2013, 77, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Booker, S.A.; Vida, I. Morphological diversity and connectivity of hippocampal interneurons. Cell Tissue Res. 2018, 373, 619–641. [Google Scholar] [CrossRef]
- Buzsaki, G. Rhythms of the Brain; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef]
- Pozo, K.; Goda, Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 2010, 66, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, D.; Carvalho, A.L. Mechanisms of homeostatic plasticity in the excitatory synapse. J. Neurochem. 2016, 139, 973–996. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Tseeb, V.; Raggozzino, D.; Khazipov, R.; Gaiarsa, J.L. gamma-Aminobutyric acid (GABA): A fast excitatory transmitter which may regulate the development of hippocampal neurones in early postnatal life. Prog. Brain Res. 1994, 102, 261–273. [Google Scholar] [CrossRef]
- Danglot, L.; Triller, A.; Marty, S. The development of hippocampal interneurons in rodents. Hippocampus 2006, 16, 1032–1060. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Kamboj, S.; Ehlers, M.D.; Rosen, K.R.; Fischbach, G.D.; Huganir, R.L. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 1998, 21, 1067–1078. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef]
- Burrone, J.; O’Byrne, M.; Murthy, V.N. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 2002, 420, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Deco, G.; Ponce-Alvarez, A.; Hagmann, P.; Romani, G.L.; Mantini, D.; Corbetta, M. How local excitation-inhibition ratio impacts the whole brain dynamics. J. Neurosci. 2014, 34, 7886–7898. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.W.; Li, Y.T.; Zhang, L.I. Formation of excitation-inhibition balance: Inhibition listens and changes its tune. Trends Neurosci. 2014, 37, 528–530. [Google Scholar] [CrossRef]
- He, H.Y.; Shen, W.; Hiramoto, M.; Cline, H.T. Experience-Dependent Bimodal Plasticity of Inhibitory Neurons in Early Development. Neuron 2016, 90, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Malinovskaya, N.A.; Komleva, Y.K.; Gorina, Y.V.; Shuvaev, A.N.; Olovyannikova, R.Y.; Belozor, O.S.; Belova, O.A.; Higashida, H.; Salmina, A.B. Excitation/inhibition imbalance and impaired neurogenesis in neurodevelopmental and neurodegenerative disorders. Rev. Neurosci. 2019, 30, 807–820. [Google Scholar] [CrossRef]
- Medendorp, W.E.; Bjorefeldt, A.; Crespo, E.L.; Prakash, M.; Pal, A.; Waddell, M.L.; Moore, C.I.; Hochgeschwender, U. Selective postnatal excitation of neocortical pyramidal neurons results in distinctive behavioral and circuit deficits in adulthood. iScience 2021, 24, 102157. [Google Scholar] [CrossRef]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef]
- Lozovaya, N.; Gataullina, S.; Tsintsadze, T.; Tsintsadze, V.; Pallesi-Pocachard, E.; Minlebaev, M.; Goriounova, N.A.; Buhler, E.; Watrin, F.; Shityakov, S.; et al. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat. Commun. 2014, 5, 4563. [Google Scholar] [CrossRef]
- He, H.Y.; Cline, H.T. What Is Excitation/Inhibition and How Is It Regulated? A Case of the Elephant and the Wisemen. J. Exp. Neurosci. 2019, 13, 1179069519859371. [Google Scholar] [CrossRef] [PubMed]
- He, H.Y.; Shen, W.; Zheng, L.; Guo, X.; Cline, H.T. Excitatory synaptic dysfunction cell-autonomously decreases inhibitory inputs and disrupts structural and functional plasticity. Nat. Commun. 2018, 9, 2893. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; McKeown, C.R.; Demas, J.A.; Cline, H.T. Inhibition to excitation ratio regulates visual system responses and behavior in vivo. J. Neurophysiol. 2011, 106, 2285–2302. [Google Scholar] [CrossRef]
- Dorrn, A.L.; Yuan, K.; Barker, A.J.; Schreiner, C.E.; Froemke, R.C. Developmental sensory experience balances cortical excitation and inhibition. Nature 2010, 465, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Huang, Z.J.; Morales, B.; Kirkwood, A. Maturation of GABAergic transmission and the timing of plasticity in visual cortex. Brain Res. Brain Res. Rev. 2005, 50, 126–133. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, L.I.; Tao, H.W. Heterosynaptic scaling of developing GABAergic synapses: Dependence on glutamatergic input and developmental stage. J. Neurosci. 2007, 27, 5301–5312. [Google Scholar] [CrossRef]
- Bassetti, D.; Lombardi, A.; Kirischuk, S.; Luhmann, H.J. Haploinsufficiency of Tsc2 Leads to Hyperexcitability of Medial Prefrontal Cortex via Weakening of Tonic GABAB Receptor-mediated Inhibition. Cereb. Cortex 2020, 30, 6313–6324. [Google Scholar] [CrossRef]
- Kramvis, I.; van Westen, R.; Lammertse, H.C.A.; Riga, D.; Heistek, T.S.; Loebel, A.; Spijker, S.; Mansvelder, H.D.; Meredith, R.M. Dysregulated Prefrontal Cortex Inhibition in Prepubescent and Adolescent Fragile X Mouse Model. Front. Mol. Neurosci. 2020, 13, 88. [Google Scholar] [CrossRef]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef]
- Bódi, V.; Májer, T.; Kelemen, V.; Világi, I.; Szűcs, A.; Varró, P. Alterations of the Hippocampal Networks in Valproic Acid-Induced Rat Autism Model. Front. Neural Circuits 2022, 16, 772792. [Google Scholar] [CrossRef]
- Antoine, M.W.; Langberg, T.; Schnepel, P.; Feldman, D.E. Increased Excitation-Inhibition Ratio Stabilizes Synapse and Circuit Excitability in Four Autism Mouse Models. Neuron 2019, 101, 648–661.e4. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.M. Epilepsy, E/I Balance and GABA(A) Receptor Plasticity. Front. Mol. Neurosci. 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Engel, J., Jr. Concepts of epilepsy. Epilepsia 1995, 36 (Suppl. S1), S23–S29. [Google Scholar] [CrossRef]
- Sloviter, R.S. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann. Neurol. 1994, 35, 640–654. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef]
- Walker, M.; Chan, D.; Thom, M. Hippocampus and human disease. In The Hippocampus Book; Andersen, P., Morris, R., Amaral, D., Bliss, T., O’Keefe, J., Eds.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Navidhamidi, M.; Ghasemi, M.; Mehranfard, N. Epilepsy-associated alterations in hippocampal excitability. Rev. Neurosci. 2017, 28, 307–334. [Google Scholar] [CrossRef]
- Avoli, M.; Louvel, J.; Pumain, R.; Köhling, R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog. Neurobiol. 2005, 77, 166–200. [Google Scholar] [CrossRef]
- Jiruska, P.; Finnerty, G.T.; Powell, A.D.; Lofti, N.; Cmejla, R.; Jefferys, J.G.R. Epileptic high-frequency network activity in a model of non-lesional temporal lobe epilepsy. Brain 2010, 133, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar] [CrossRef]
- Mulley, J.C.; Scheffer, I.E.; Harkin, L.A.; Berkovic, S.F.; Dibbens, L.M. Susceptibility genes for complex epilepsy. Human. Mol. Genet. 2005, 14, R243–R249. [Google Scholar] [CrossRef]
- Žiburkus, J.; Cressman, J.R.; Schiff, S.J. Seizures as imbalanced up states: Excitatory and inhibitory conductances during seizure-like events. J. Neurophysiol. 2013, 109, 1296–1306. [Google Scholar] [CrossRef]
- Toth, K.; Hofer, K.T.; Kandracs, A.; Entz, L.; Bago, A.; Eross, L.; Jordan, Z.; Nagy, G.; Solyom, A.; Fabo, D.; et al. Hyperexcitability of the network contributes to synchronization processes in the human epileptic neocortex. J. Physiol. 2017, 596, 317–342. [Google Scholar] [CrossRef]
- Duma, G.M.; Cuozzo, S.; Wilson, L.; Danieli, A.; Bonanni, P.; Pellegrino, G. Excitation/Inhibition balance relates to cognitive function and gene expression in temporal lobe epilepsy: A high density EEG assessment with aperiodic exponent. Brain Commun. 2024, 6, fcae231. [Google Scholar] [CrossRef] [PubMed]
- Paluszkiewicz, S.M.; Martin, B.S.; Huntsman, M.M. Fragile X syndrome: The GABAergic system and circuit dysfunction. Dev. Neurosci. 2011, 33, 349–364. [Google Scholar] [CrossRef]
- Nomura, T. Interneuron Dysfunction and Inhibitory Deficits in Autism and Fragile X Syndrome. Cells 2021, 10, 2610. [Google Scholar] [CrossRef] [PubMed]
- Morin-Parent, F.; Champigny, C.; Lacroix, A.; Corbin, F.; Lepage, J.F. Hyperexcitability and impaired intracortical inhibition in patients with fragile-X syndrome. Transl. Psychiatry 2019, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Svalina, M.N.; Guthman, E.M.; Cea-Del Rio, C.A.; Kushner, J.K.; Baca, S.M.; Restrepo, D.; Huntsman, M.M. Hyperexcitability and Loss of Feedforward Inhibition Contribute to Aberrant Plasticity in the Fmr1KO Amygdala. eNeuro 2021, 8. [Google Scholar] [CrossRef]
- Lányi, O.; Koleszár, B.; Schulze Wenning, A.; Balogh, D.; Engh, M.A.; Horváth, A.A.; Fehérvari, P.; Hegyi, P.; Molnár, Z.; Unoka, Z.; et al. Excitation/inhibition imbalance in schizophrenia: A meta-analysis of inhibitory and excitatory TMS-EMG paradigms. Schizophrenia 2024, 10, 56. [Google Scholar] [CrossRef]
- Liu, Y.; Ouyang, P.; Zheng, Y.; Mi, L.; Zhao, J.; Ning, Y.; Guo, W. A Selective Review of the Excitatory-Inhibitory Imbalance in Schizophrenia: Underlying Biology, Genetics, Microcircuits, and Symptoms. Front. Cell Dev. Biol. 2021, 9, 664535. [Google Scholar] [CrossRef]
- Hu, Y.T.; Tan, Z.L.; Hirjak, D.; Northoff, G. Brain-wide changes in excitation-inhibition balance of major depressive disorder: A systematic review of topographic patterns of GABA- and glutamatergic alterations. Mol. Psychiatry 2023, 28, 3257–3266. [Google Scholar] [CrossRef]
- Bartsch, T.; Wulff, P. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience 2015, 309, 1–16. [Google Scholar] [CrossRef]
- Geuze, E.; Vermetten, E.; Bremner, J.D. MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol. Psychiatry 2005, 10, 160–184. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T. (Ed.) Introduction: The hippocampus in the clinical neurosciences. In The Clinical Neurobiology of the Hippocampus: An Integrative View; Oxford University Press (Oxford Scholarship Online): Oxford, UK, 2012. [Google Scholar] [CrossRef]
- Peyton, L.; Oliveros, A.; Choi, D.S.; Jang, M.H. Hippocampal regenerative medicine: Neurogenic implications for addiction and mental disorders. Exp. Mol. Med. 2021, 53, 358–368. [Google Scholar] [CrossRef]
- Sapolsky, R.M. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry 2000, 57, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Schwartzkroin, P.A. Role of the hippocampus in epilepsy. Hippocampus 1994, 4, 239–242. [Google Scholar] [CrossRef]
- Noebels, J.L.; Avoli, M.; Rogawski, M.; Olsen, R.; Delgado-Escueta, A.V. Jasper’s Basic Mechanisms of the Epilepsies; OUP USA: Oxford, UK, 2012; Volume 80. [Google Scholar]
- Barker-Haliski, M.; White, H.S. Glutamatergic Mechanisms Associated with Seizures and Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, a022863. [Google Scholar] [CrossRef] [PubMed]
- Kleen, J.K.; Scott, R.C.; Holmes, G.L.; Roberts, D.W.; Rundle, M.M.; Testorf, M.; Lenck-Santini, P.P.; Jobst, B.C. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 2013, 81, 18–24. [Google Scholar] [CrossRef]
- Jung, R. Hirnelektrische Untersuchungen über den Elektrokrampf: Die Erregungsabläufe in corticalen und subcorticalen Hirnregionen bei Katze und Hund. Arch. Für Psychiatr. Und Nervenkrankh. 1949, 183, 206–244. [Google Scholar] [CrossRef]
- Liberson, W.T.; Akert, K. Hippocampal seizure states in guinea pig. Electroencephalogr. Clin. Neurophysiol. 1955, 7, 211–222. [Google Scholar]
- Green, J.D.; Shimamoto, T. Hippocampal seizures and their propagation. Arch. Neurol. Psychiatry 1953, 70, 687–702. [Google Scholar] [CrossRef]
- Green, J.D. The Hippocampus. Physiol. Rev. 1964, 44, 561–608. [Google Scholar] [CrossRef] [PubMed]
- Andy, O.J.; Akert, K. Seizure patterns induced by electrical stimulation of hippocampal formation in the cat. J. Neuropathol. Exp. Neurol. 1955, 14, 198–213. [Google Scholar] [PubMed]
- Chatzikonstantinou, A. Epilepsy and the hippocampus. Front. Neurol. Neurosci. 2014, 34, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Benbadis, S.R. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology 1999, 53, 440. [Google Scholar] [CrossRef]
- Engel, J., Jr. Mesial temporal lobe epilepsy: What have we learned? Neuroscientist 2001, 7, 340–352. [Google Scholar] [CrossRef]
- Wiebe, S. Epidemiology of temporal lobe epilepsy. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 2000, 27 (Suppl. S1), S6–S10; discussion S20-11. [Google Scholar] [CrossRef]
- King, D.; Bronen, R.A.; Spencer, D.D.; Spencer, S.S. Topographic distribution of seizure onset and hippocampal atrophy: Relationship between MRI and depth EEG. Electroencephalogr. Clin. Neurophysiol. 1997, 103, 692–697. [Google Scholar]
- Bertram, E.H. Temporal lobe epilepsy: Where do the seizures really begin? Epilepsy Behav. EB 2009, 14 (Suppl. S1), 32–37. [Google Scholar] [CrossRef]
- Leong, E.C.S.; Seneviratne, U. “Benign” temporal lobe epilepsy with hippocampal sclerosis: A forgotten entity? Epilepsy Behav. Rep. 2020, 14, 100407. [Google Scholar] [CrossRef]
- Jefferys, J.G. Hippocampal sclerosis and temporal lobe epilepsy: Cause or consequence? Brain 1999, 122 Pt 6, 1007–1008. [Google Scholar] [CrossRef]
- Burnham, W.M. Primary and “transfer” seizure development in the kindled rat. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 1975, 2, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Lothman, E.W.; Collins, R.C. Kainic acid induced limbic seizures: Metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981, 218, 299–318. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, D.C.; Nathanson, D.; Edson, N. A new model of partial status epilepticus based on kindling. Brain Res. 1982, 250, 53–63. [Google Scholar] [CrossRef]
- Becker, A.; Letzel, K.; Letzel, U.; Grecksch, G. Kindling of the dorsal and the ventral hippocampus: Effects on learning performance in rats. Physiol. Behav. 1997, 62, 1265–1271. [Google Scholar] [CrossRef]
- Akaike, K.; Tanaka, S.; Tojo, H.; Fukumoto, S.; Imamura, S.; Takigawa, M. Kainic acid-induced dorsal and ventral hippocampal seizures in rats. Brain Res. 2001, 900, 65–71. [Google Scholar] [CrossRef]
- Haussler, U.; Bielefeld, L.; Froriep, U.P.; Wolfart, J.; Haas, C.A. Septotemporal position in the hippocampal formation determines epileptic and neurogenic activity in temporal lobe epilepsy. Cereb. Cortex 2012, 22, 26–36. [Google Scholar] [CrossRef]
- Greco, B.; Prevost, J.; Gioanni, Y. Intracerebral microinjections of dermorphin: Search for the epileptic induction thresholds. Neuroreport 1994, 5, 2169–2172. [Google Scholar] [CrossRef]
- Racine, R.; Rose, P.A.; Burnham, W.M. Afterdischarge thresholds and kindling rates in dorsal and ventral hippocampus and dentate gyrus. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 1977, 4, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Apland, J.P.; Figueiredo, T.H.; Qashu, F.; Aroniadou-Anderjaska, V.; Souza, A.P.; Braga, M.F. Higher susceptibility of the ventral versus the dorsal hippocampus and the posteroventral versus anterodorsal amygdala to soman-induced neuropathology. Neurotoxicology 2010, 31, 485–492. [Google Scholar] [CrossRef]
- Gilbert, M.; Racine, R.J.; Smith, G.K. Epileptiform burst responses in ventral vs dorsal hippocampal slices. Brain Res. 1985, 361, 389–391. [Google Scholar] [CrossRef]
- Bragdon, A.C.; Taylor, D.M.; Wilson, W.A. Potassium-induced epileptiform activity in area CA3 varies markedly along the septotemporal axis of the rat hippocampus. Brain Res. 1986, 378, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Xie, C.W.; Lewis, D.V.; Wilson, W.A.; Mitchell, C.L.; Hong, J.S. Opioid-induced epileptiform bursting in hippocampal slices: Higher susceptibility in ventral than dorsal hippocampus. J. Pharmacol. Exp. Ther. 1990, 253, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Borck, C.; Jefferys, J.G. Seizure-like events in disinhibited ventral slices of adult rat hippocampus. J. Neurophysiol. 1999, 82, 2130–2142. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoropoulos, C.; Moschovos, C.; Kostopoulos, G. Greater contribution of N-methyl-D-aspartic acid receptors in ventral compared to dorsal hippocampal slices in the expression and long-term maintenance of epileptiform activity. Neuroscience 2005, 135, 765–779. [Google Scholar] [CrossRef]
- Moschovos, C.; Kostopoulos, G.; Papatheodoropoulos, C. Endogenous adenosine induces NMDA receptor-independent persistent epileptiform discharges in dorsal and ventral hippocampus via activation of A2 receptors. Epilepsy Res. 2012, 100, 157–167. [Google Scholar] [CrossRef]
- Papatheodoropoulos, C. Higher intrinsic network excitability in ventral compared with the dorsal hippocampus is controlled less effectively by GABAB receptors. BMC Neurosci. 2015, 16, 75. [Google Scholar] [CrossRef]
- Mikroulis, A.V.; Psarropoulou, C. Endogenous ACh effects on NMDA-induced interictal-like discharges along the septotemporal hippocampal axis of adult rats and their modulation by an early life generalized seizure. Epilepsia 2012, 53, 879–887. [Google Scholar] [CrossRef]
- Dzhala, V.; Khalilov, I.; Ben-Ari, Y.; Khazipov, R. Neuronal mechanisms of the anoxia-induced network oscillations in the rat hippocampus in vitro. J. Physiol. 2001, 536, 521–531. [Google Scholar] [CrossRef]
- Derchansky, M.; Shahar, E.; Wennberg, R.A.; Samoilova, M.; Jahromi, S.S.; Abdelmalik, P.A.; Zhang, L.; Carlen, P.L. Model of frequent, recurrent, and spontaneous seizures in the intact mouse hippocampus. Hippocampus 2004, 14, 935–947. [Google Scholar] [CrossRef]
- Buckmaster, P.S.; Reyes, B.; Kahn, T.; Wyeth, M. Ventral Hippocampal Formation Is the Primary Epileptogenic Zone in a Rat Model of Temporal Lobe Epilepsy. J. Neurosci. 2022, 42, 7482–7495. [Google Scholar] [CrossRef]
- Isaeva, E.; Romanov, A.; Holmes, G.L.; Isaev, D. Status epilepticus results in region-specific alterations in seizure susceptibility along the hippocampal longitudinal axis. Epilepsy Res. 2015, 110, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Pikkarainen, M.; Ronkko, S.; Savander, V.; Insausti, R.; Pitkanen, A. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the hippocampal formation in rat. J. Comp. Neurol. 1999, 403, 229–260. [Google Scholar]
- Pitkanen, A.; Pikkarainen, M.; Nurminen, N.; Ylinen, A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann. N. Y. Acad. Sci. 2000, 911, 369–391. [Google Scholar]
- Aroniadou-Anderjaska, V.; Fritsch, B.; Qashu, F.; Braga, M.F. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008, 78, 102–116. [Google Scholar] [CrossRef]
- Makhalova, J.; Le Troter, A.; Aubert-Conil, S.; Giusiano, B.; McGonigal, A.; Trebuchon, A.; Carron, R.; Medina Villalon, S.; Bénar, C.G.; Ranjeva, J.P.; et al. Epileptogenic networks in drug-resistant epilepsy with amygdala enlargement: Assessment with stereo-EEG and 7 T MRI. Clin. Neurophysiol. 2022, 133, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Agster, K.L.; Burwell, R.D. Hippocampal and subicular efferents and afferents of the perirhinal, postrhinal, and entorhinal cortices of the rat. Behav. Brain Res. 2013, 254, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, Y.; Peng, J.; Zhao, Y.; He, X.; Yu, X.; Liu, Q.; Jin, S.; Xu, F. Whole-Brain Mapping the Direct Inputs of Dorsal and Ventral CA1 Projection Neurons. Front. Neural Circuits 2021, 15, 643230. [Google Scholar] [CrossRef]
- Witter, M.P.; Groenewegen, H.J.; Lopes da Silva, F.H.; Lohman, A.H. Functional organization of the extrinsic and intrinsic circuitry of the parahippocampal region. Prog. Neurobiol. 1989, 33, 161–253. [Google Scholar]
- Wozny, C.; Gabriel, S.; Jandova, K.; Schulze, K.; Heinemann, U.; Behr, J. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol. Dis. 2005, 19, 451–460. [Google Scholar] [CrossRef]
- Ang, C.W.; Carlson, G.C.; Coulter, D.A. Massive and specific dysregulation of direct cortical input to the hippocampus in temporal lobe epilepsy. J. Neurosci. 2006, 26, 11850–11856. [Google Scholar] [CrossRef]
- Toyoda, I.; Bower, M.R.; Leyva, F.; Buckmaster, P.S. Early activation of ventral hippocampus and subiculum during spontaneous seizures in a rat model of temporal lobe epilepsy. J. Neurosci. 2013, 33, 11100–11115. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Diespirov, G.P.; Malkin, S.L.; Chernyshev, A.S.; Vylekzhanina, E.N.; Zaitsev, A.V. Morphological and Functional Alterations in the CA1 Pyramidal Neurons of the Rat Hippocampus in the Chronic Phase of the Lithium-Pilocarpine Model of Epilepsy. Int. J. Mol. Sci. 2024, 25, 7568. [Google Scholar] [CrossRef]
- Rashid, S.; Pho, G.; Czigler, M.; Werz, M.A.; Durand, D.M. Low frequency stimulation of ventral hippocampal commissures reduces seizures in a rat model of chronic temporal lobe epilepsy. Epilepsia 2012, 53, 147–156. [Google Scholar] [CrossRef]
- Zeidler, Z.; Brandt-Fontaine, M.; Leintz, C.; Krook-Magnuson, C.; Netoff, T.; Krook-Magnuson, E. Targeting the Mouse Ventral Hippocampus in the Intrahippocampal Kainic Acid Model of Temporal Lobe Epilepsy. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Li, M.; Jiang, Y.Q.; Lee, D.K.; Wang, H.; Lu, M.C.; Sun, Q. Dorsoventral Heterogeneity of Synaptic Connectivity in Hippocampal CA3 Pyramidal Neurons. J. Neurosci. 2024, 44, e0370242024. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, K.A.; Islam, T.; Johnston, D. Intrinsic excitability of CA1 pyramidal neurones from the rat dorsal and ventral hippocampus. J. Physiol. 2012, 590, 5707–5722. [Google Scholar] [CrossRef]
- Dougherty, K.A.; Nicholson, D.A.; Diaz, L.; Buss, E.W.; Neuman, K.M.; Chetkovich, D.M.; Johnston, D. Differential expression of HCN subunits alters voltage-dependent gating of h-channels in CA1 pyramidal neurons from dorsal and ventral hippocampus. J. Neurophysiol. 2013, 109, 1940–1953. [Google Scholar] [CrossRef] [PubMed]
- Honigsperger, C.; Marosi, M.; Murphy, R.; Storm, J.F. Dorsoventral differences in Kv7/M-current and its impact on resonance, temporal summation and excitability in rat hippocampal pyramidal cells. J. Physiol. 2015, 593, 1551–1580. [Google Scholar] [CrossRef]
- Malik, R.; Dougherty, K.A.; Parikh, K.; Byrne, C.; Johnston, D. Mapping the electrophysiological and morphological properties of CA1 pyramidal neurons along the longitudinal hippocampal axis. Hippocampus 2016, 26, 341–361. [Google Scholar] [CrossRef]
- Milior, G.; Castro, M.A.; Sciarria, L.P.; Garofalo, S.; Branchi, I.; Ragozzino, D.; Limatola, C.; Maggi, L. Electrophysiological Properties of CA1 Pyramidal Neurons along the Longitudinal Axis of the Mouse Hippocampus. Sci. Rep. 2016, 6, 38242. [Google Scholar] [CrossRef]
- Trompoukis, G.; Leontiadis, L.J.; Rigas, P.; Papatheodoropoulos, C. Scaling of Network Excitability and Inhibition may Contribute to the Septotemporal Differentiation of Sharp Waves-Ripples in Rat Hippocampus In Vitro. Neuroscience 2021, 458, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Kouvaros, S.; Papatheodoropoulos, C. Major dorsoventral differences in the modulation of the local CA1 hippocampal network by NMDA, mGlu5, adenosine A2A and cannabinoid CB1 receptors. Neuroscience 2016, 317, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Stöber, T.M.; Batulin, D.; Triesch, J.; Narayanan, R.; Jedlicka, P. Degeneracy in epilepsy: Multiple routes to hyperexcitable brain circuits and their repair. Commun. Biol. 2023, 6, 479. [Google Scholar] [CrossRef]
- Derchansky, M.; Rokni, D.; Rick, J.T.; Wennberg, R.; Bardakjian, B.L.; Zhang, L.; Yarom, Y.; Carlen, P.L. Bidirectional multisite seizure propagation in the intact isolated hippocampus: The multifocality of the seizure “focus”. Neurobiol. Dis. 2006, 23, 312–328. [Google Scholar] [CrossRef]
- Marx, M.; Haas, C.A.; Haussler, U. Differential vulnerability of interneurons in the epileptic hippocampus. Front. Cell. Neurosci. 2013, 7, 167. [Google Scholar] [CrossRef]
- Murthy, S.; Kane, G.A.; Katchur, N.J.; Lara Mejia, P.S.; Obiofuma, G.; Buschman, T.J.; McEwen, B.S.; Gould, E. Perineuronal Nets, Inhibitory Interneurons, and Anxiety-Related Ventral Hippocampal Neuronal Oscillations Are Altered by Early Life Adversity. Biol. Psychiatry 2019, 85, 1011–1020. [Google Scholar] [CrossRef]
- Smeralda, C.L.; Pandit, S.; Turrini, S.; Reilly, J.; Palmisano, A.; Sprugnoli, G.; Hampel, H.; Benussi, A.; Borroni, B.; Press, D.; et al. The role of parvalbumin interneuron dysfunction across neurodegenerative dementias. Ageing Res. Rev. 2024, 101, 102509. [Google Scholar] [CrossRef]
- Leitch, B. Parvalbumin Interneuron Dysfunction in Neurological Disorders: Focus on Epilepsy and Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 5549. [Google Scholar] [CrossRef]
- Moxon, K.A.; Shahlaie, K.; Girgis, F.; Saez, I.; Kennedy, J.; Gurkoff, G.G. From adagio to allegretto: The changing tempo of theta frequencies in epilepsy and its relation to interneuron function. Neurobiol. Dis. 2019, 129, 169–181. [Google Scholar] [CrossRef]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 2007, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, K.; Kanodia, H.; Donato, F.; Caroni, P. Selective vulnerability of the ventral hippocampus-prelimbic cortex axis parvalbumin interneuron network underlies learning deficits of fragile X mice. Cell Rep. 2024, 43, 114124. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoropoulos, C.; Asprodini, E.; Nikita, I.; Koutsona, C.; Kostopoulos, G. Weaker synaptic inhibition in CA1 region of ventral compared to dorsal rat hippocampal slices. Brain Res. 2002, 948, 117–121. [Google Scholar] [CrossRef]
- Petrides, T.; Georgopoulos, P.; Kostopoulos, G.; Papatheodoropoulos, C. The GABAA receptor-mediated recurrent inhibition in ventral compared with dorsal CA1 hippocampal region is weaker, decays faster and lasts less. Exp. Brain Res. 2007, 177, 370–383. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Differential corticosteroid modulation of inhibitory synaptic currents in the dorsal and ventral hippocampus. J. Neurosci. 2009, 29, 2857–2866. [Google Scholar] [CrossRef]
- Valero-Aracama, M.J.; Zheng, F.; Alzheimer, C. Dorsal-Ventral Gradient of Activin Regulates Strength of GABAergic Inhibition along Longitudinal Axis of Mouse Hippocampus in an Activity-Dependent Fashion. Int. J. Mol. Sci. 2023, 24, 13145. [Google Scholar] [CrossRef] [PubMed]
- Pofantis, H.; Georgopoulos, P.; Petrides, T.; Papatheodoropoulos, C. Differences in paired-pulse inhibition and facilitation in the dentate gyrus and CA3 field between dorsal and ventral rat hippocampus. Brain Res. 2015, 1608, 21–30. [Google Scholar] [CrossRef]
- Schreurs, A.; Sabanov, V.; Balschun, D. Distinct Properties of Long-Term Potentiation in the Dentate Gyrus along the Dorsoventral Axis: Influence of Age and Inhibition. Sci. Rep. 2017, 7, 5157. [Google Scholar] [CrossRef]
- Lopez-Santiago, L.F.; Yuan, Y.; Wagnon, J.L.; Hull, J.M.; Frasier, C.R.; O’Malley, H.A.; Meisler, M.H.; Isom, L.L. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 2383–2388. [Google Scholar] [CrossRef]
- Hofmann, G.; Balgooyen, L.; Mattis, J.; Deisseroth, K.; Buckmaster, P.S. Hilar somatostatin interneuron loss reduces dentate gyrus inhibition in a mouse model of temporal lobe epilepsy. Epilepsia 2016, 57, 977–983. [Google Scholar] [CrossRef]
- Wittner, L.; Magloczky, Z. Synaptic Reorganization of the Perisomatic Inhibitory Network in Hippocampi of Temporal Lobe Epileptic Patients. BioMed Res. Int. 2017, 2017, 7154295. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S.; Zappone, C.A.; Harvey, B.D.; Frotscher, M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: Possible anatomical substrate of granule cell hyper-inhibition in chronically epileptic rats. J. Comp. Neurol. 2006, 494, 944–960. [Google Scholar] [CrossRef] [PubMed]
- Dengler, C.G.; Coulter, D.A. Normal and epilepsy-associated pathologic function of the dentate gyrus. Prog. Brain Res. 2016, 226, 155–178. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, E.J.; Gupta, A.; Subramanian, D.; Korgaonkar, A.A.; Santhakumar, V. Converging early responses to brain injury pave the road to epileptogenesis. J. Neurosci. Res. 2019, 97, 1335–1344. [Google Scholar] [CrossRef]
- Milton, C.K.; O’Neal, C.M.; Conner, A.K. Functional connectivity of hippocampus in temporal lobe epilepsy depends on hippocampal dominance: A systematic review of the literature. J. Neurol. 2022, 269, 221–232. [Google Scholar] [CrossRef]
- Scheibel, M.E.; Crandall, P.H.; Scheibel, A.B. The hippocampal-dentate complex in temporal lobe epilepsy. A Golgi study. Epilepsia 1974, 15, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Schwarzer, C.; Lothman, E.W.; Williamson, J.; Sperk, G. Functional changes in somatostatin and neuropeptide Y containing neurons in the rat hippocampus in chronic models of limbic seizures. Epilepsy Res. 1996, 26, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Elmér, E.; Kokaia, M.; Kokaia, Z.; Ferencz, I.; Lindvall, O. Delayed kindling development after rapidly recurring seizures: Relation to mossy fiber sprouting and neurotrophin, GAP-43 and dynorphin gene expression. Brain Res. 1996, 712, 19–34. [Google Scholar] [CrossRef]
- Lothman, E.W.; Williamson, J.M. Rapid kindling with recurrent hippocampal seizures. Epilepsy Res. 1993, 14, 209–220. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Y.; Cheng, H.; Li, Z.; Lai, N.; Li, M.; Ruan, Y.; Zheng, Y.; Fei, F.; Xu, C.; et al. Adult-born neurons in critical period maintain hippocampal seizures via local aberrant excitatory circuits. Signal Transduct. Target. Ther. 2023, 8, 225. [Google Scholar] [CrossRef]
- Sloviter, R.S. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science 1987, 235, 73–76. [Google Scholar] [CrossRef]
- Kobayashi, M.; Buckmaster, P.S. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J. Neurosci. 2003, 23, 2440–2452. [Google Scholar] [CrossRef] [PubMed]
- Avoli, M.; de Curtis, M.; Gnatkovsky, V.; Gotman, J.; Köhling, R.; Lévesque, M.; Manseau, F.; Shiri, Z.; Williams, S. Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J. Neurophysiol. 2016, 115, 3229–3237. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: The “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus 1991, 1, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Andrioli, A.; Alonso-Nanclares, L.; Arellano, J.I.; DeFelipe, J. Quantitative analysis of parvalbumin-immunoreactive cells in the human epileptic hippocampus. Neuroscience 2007, 149, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gan, J.; Jonas, P. Interneurons. Fast-spiking, parvalbumin⁺ GABAergic interneurons: From cellular design to microcircuit function. Science 2014, 345, 1255263. [Google Scholar] [CrossRef]
- Sperk, G.; Wieselthaler-Hölzl, A.; Pirker, S.; Tasan, R.; Strasser, S.S.; Drexel, M.; Pifl, C.; Marschalek, J.; Ortler, M.; Trinka, E.; et al. Glutamate decarboxylase 67 is expressed in hippocampal mossy fibers of temporal lobe epilepsy patients. Hippocampus 2012, 22, 590–603. [Google Scholar] [CrossRef]
- Vezzani, A.; Michalkiewicz, M.; Michalkiewicz, T.; Moneta, D.; Ravizza, T.; Richichi, C.; Aliprandi, M.; Mulé, F.; Pirona, L.; Gobbi, M.; et al. Seizure susceptibility and epileptogenesis are decreased in transgenic rats overexpressing neuropeptide Y. Neuroscience 2002, 110, 237–243. [Google Scholar] [CrossRef]
- Walker, M.C.; Kullmann, D.M. Tonic GABA(A) Receptor-Mediated Signaling in Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US) Copyright © 2012: Bethesda, MD, USA, 2012. [Google Scholar]
- Li, Z.X.; Yu, H.M.; Jiang, K.W. Tonic GABA inhibition in hippocampal dentate granule cells: Its regulation and function in temporal lobe epilepsies. Acta Physiol. 2013, 209, 199–211. [Google Scholar] [CrossRef]
- Chancey, J.H.; Howard, M.A. Synaptic Integration in CA1 Pyramidal Neurons Is Intact despite Deficits in GABAergic Transmission in the Scn1a Haploinsufficiency Mouse Model of Dravet Syndrome. eNeuro 2022, 9. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Bassell, G.J.; Warren, S.T. Fragile X syndrome: Loss of local mRNA regulation alters synaptic development and function. Neuron 2008, 60, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Kooy, R.F.; D’Hooge, R.; Reyniers, E.; Bakker, C.E.; Nagels, G.; De Boulle, K.; Storm, K.; Clincke, G.; De Deyn, P.P.; Oostra, B.A.; et al. Transgenic mouse model for the fragile X syndrome. Am. J. Med. Genet. 1996, 64, 241–245. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Kidd, S.A.; Lachiewicz, A.; Barbouth, D.; Blitz, R.K.; Delahunty, C.; McBrien, D.; Visootsak, J.; Berry-Kravis, E. Fragile X syndrome: A review of associated medical problems. Pediatrics 2014, 134, 995–1005. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139, S194–S206. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Mesibov, G.B.; Hatton, D.D.; Clark, R.D.; Roberts, J.E.; Mayhew, L. Autistic behavior in young boys with fragile X syndrome. J. Autism Dev. Disord. 1998, 28, 499–508. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Jackson, A.W., 3rd; Levitas, A.; Rimland, B.; Braden, M. An analysis of autism in fifty males with the fragile X syndrome. Am. J. Med. Genet. 1986, 23, 359–374. [Google Scholar] [CrossRef]
- Belmonte, M.K.; Bourgeron, T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat. Neurosci. 2006, 9, 1221–1225. [Google Scholar] [CrossRef]
- Berry-Kravis, E. Epilepsy in fragile X syndrome. Dev. Med. Child Neurol. 2002, 44, 724–728. [Google Scholar] [CrossRef]
- Incorpora, G.; Sorge, G.; Sorge, A.; Pavone, L. Epilepsy in fragile X syndrome. Brain Dev. 2002, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kluger, G.; Böhm, I.; Laub, M.C.; Waldenmaier, C. Epilepsy and fragile X gene mutations. Pediatr. Neurol. 1996, 15, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Filipink, R.A.; Frye, R.E.; Golla, S.; Morris, S.M.; Andrews, H.; Choo, T.H.; Kaufmann, W.E. Seizures in Fragile X Syndrome: Associations and Longitudinal Analysis of a Large Clinic-Based Cohort. Front. Pediatr. 2021, 9, 736255. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; French, J.H.; Fernando, S.; Brown, W.T.; Jenkins, E.C.; Friedman, E.; Hill, A.L.; Miezejeski, C.M. Fragile X syndrome: Associated neurological abnormalities and developmental disabilities. Ann. Neurol. 1985, 18, 665–669. [Google Scholar] [CrossRef]
- Sabaratnam, M.; Vroegop, P.G.; Gangadharan, S.K. Epilepsy and EEG findings in 18 males with fragile X syndrome. Seizure 2001, 10, 60–63. [Google Scholar] [CrossRef]
- Pfeiffer, B.E.; Huber, K.M. Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 2007, 27, 3120–3130. [Google Scholar] [CrossRef]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Booker, S.A.; Kind, P.C. Mechanisms regulating input-output function and plasticity of neurons in the absence of FMRP. Brain Res. Bull. 2021, 175, 69–80. [Google Scholar] [CrossRef]
- Aishworiya, R.; Protic, D.; Hagerman, R. Autism spectrum disorder in the fragile X premutation state: Possible mechanisms and implications. J. Neurol. 2022, 269, 4676–4683. [Google Scholar] [CrossRef]
- Long, J.; Li, H.; Liu, Y.; Liao, X.; Tang, Z.; Han, K.; Chen, J.; Zhang, H. Insights into the structure and function of the hippocampus: Implications for the pathophysiology and treatment of autism spectrum disorder. Front. Psychiatry 2024, 15, 1364858. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, J.; Gong, H.; Liu, T.; Li, X.; Fan, X. Implication of Hippocampal Neurogenesis in Autism Spectrum Disorder: Pathogenesis and Therapeutic Implications. Curr. Neuropharmacol. 2022, 21, 2266–2282. [Google Scholar] [CrossRef]
- Banker, S.M.; Gu, X.; Schiller, D.; Foss-Feig, J.H. Hippocampal contributions to social and cognitive deficits in autism spectrum disorder. Trends Neurosci. 2021, 44, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Ordemann, G.J.; Apgar, C.J.; Chitwood, R.A.; Brager, D.H. Altered A-type potassium channel function impairs dendritic spike initiation and temporoammonic long-term potentiation in Fragile X syndrome. J. Neurosci. 2021, 41, 5947–5962. [Google Scholar] [CrossRef] [PubMed]
- Gatto, C.L.; Broadie, K. The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol. Neurobiol. 2009, 39, 107–129. [Google Scholar] [CrossRef]
- D’Hooge, R.; Nagels, G.; Franck, F.; Bakker, C.E.; Reyniers, E.; Storm, K.; Kooy, R.F.; Oostra, B.A.; Willems, P.J.; De Deyn, P.P. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 1997, 76, 367–376. [Google Scholar] [CrossRef]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef]
- Gonçalves, J.T.; Anstey, J.E.; Golshani, P.; Portera-Cailliau, C. Circuit level defects in the developing neocortex of Fragile X mice. Nat. Neurosci. 2013, 16, 903–909. [Google Scholar] [CrossRef]
- Domanski, A.P.F.; Booker, S.A.; Wyllie, D.J.A.; Isaac, J.T.R.; Kind, P.C. Cellular and synaptic phenotypes lead to disrupted information processing in Fmr1-KO mouse layer 4 barrel cortex. Nat. Commun. 2019, 10, 4814. [Google Scholar] [CrossRef]
- Routh, B.N.; Rathour, R.K.; Baumgardner, M.E.; Kalmbach, B.E.; Johnston, D.; Brager, D.H. Increased transient Na(+) conductance and action potential output in layer 2/3 prefrontal cortex neurons of the fmr1(-/y) mouse. J. Physiol. 2017, 595, 4431–4448. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, Z.; Zhu, P.; Li, M.; Yi, Y.H.; Liao, W.P.; Su, T. Altered intrinsic properties and bursting activities of neurons in layer IV of somatosensory cortex from Fmr-1 knockout mice. Exp. Neurol. 2016, 280, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.Y.; Klyachko, V.A. Increased Persistent Sodium Current Causes Neuronal Hyperexcitability in the Entorhinal Cortex of Fmr1 Knockout Mice. Cell Rep. 2016, 16, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Kalmbach, B.E.; Johnston, D.; Brager, D.H. Cell-Type Specific Channelopathies in the Prefrontal Cortex of the fmr1-/y Mouse Model of Fragile X Syndrome. eNeuro 2015, 2. [Google Scholar] [CrossRef]
- Luque, M.A.; Beltran-Matas, P.; Marin, M.C.; Torres, B.; Herrero, L. Excitability is increased in hippocampal CA1 pyramidal cells of Fmr1 knockout mice. PLoS ONE 2017, 12, e0185067. [Google Scholar] [CrossRef]
- Deng, P.Y.; Carlin, D.; Oh, Y.M.; Myrick, L.K.; Warren, S.T.; Cavalli, V.; Klyachko, V.A. Voltage-Independent SK-Channel Dysfunction Causes Neuronal Hyperexcitability in the Hippocampus of Fmr1 Knock-Out Mice. J. Neurosci. 2019, 39, 28–43. [Google Scholar] [CrossRef]
- Chuang, S.C.; Zhao, W.; Bauchwitz, R.; Yan, Q.; Bianchi, R.; Wong, R.K. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci. 2005, 25, 8048–8055. [Google Scholar] [CrossRef]
- Booker, S.A.; Domanski, A.P.F.; Dando, O.R.; Jackson, A.D.; Isaac, J.T.R.; Hardingham, G.E.; Wyllie, D.J.A.; Kind, P.C. Altered dendritic spine function and integration in a mouse model of fragile X syndrome. Nat. Commun. 2019, 10, 4813. [Google Scholar] [CrossRef]
- Gildin, L.; Rauti, R.; Vardi, O.; Kuznitsov-Yanovsky, L.; Maoz, B.M.; Segal, M.; Ben-Yosef, D. Impaired Functional Connectivity Underlies Fragile X Syndrome. Int. J. Mol. Sci. 2022, 23, 2048. [Google Scholar] [CrossRef]
- Deng, P.Y.; Avraham, O.; Cavalli, V.; Klyachko, V.A. Hyperexcitability of Sensory Neurons in Fragile X Mouse Model. Front. Mol. Neurosci. 2021, 14, 796053. [Google Scholar] [CrossRef]
- Boone, C.E.; Davoudi, H.; Harrold, J.B.; Foster, D.J. Abnormal Sleep Architecture and Hippocampal Circuit Dysfunction in a Mouse Model of Fragile X Syndrome. Neuroscience 2018, 384, 275–289. [Google Scholar] [CrossRef]
- Contractor, A.; Klyachko, V.A.; Portera-Cailliau, C. Altered Neuronal and Circuit Excitability in Fragile X Syndrome. Neuron 2015, 87, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Brager, D.H.; Johnston, D. Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res. Bull. 2014, 103, 11–17. [Google Scholar] [CrossRef]
- Bhakar, A.L.; Dölen, G.; Bear, M.F. The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 2012, 35, 417–443. [Google Scholar] [CrossRef]
- Gross, C.; Yao, X.; Pong, D.L.; Jeromin, A.; Bassell, G.J. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J. Neurosci. 2011, 31, 5693–5698. [Google Scholar] [CrossRef] [PubMed]
- Brager, D.H.; Akhavan, A.R.; Johnston, D. Impaired dendritic expression and plasticity of h-channels in the fmr1(-/y) mouse model of fragile X syndrome. Cell Rep. 2012, 1, 225–233. [Google Scholar] [CrossRef]
- Zhang, Y.; Bonnan, A.; Bony, G.; Ferezou, I.; Pietropaolo, S.; Ginger, M.; Sans, N.; Rossier, J.; Oostra, B.; LeMasson, G.; et al. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1(-/y) mice. Nat. Neurosci. 2014, 17, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Brandalise, F.; Kalmbach, B.E.; Mehta, P.; Thornton, O.; Johnston, D.; Zemelman, B.V.; Brager, D.H. Fragile X Mental Retardation Protein Bidirectionally Controls Dendritic I(h) in a Cell Type-Specific Manner between Mouse Hippocampus and Prefrontal Cortex. J. Neurosci. 2020, 40, 5327–5340. [Google Scholar] [CrossRef]
- Routh, B.N.; Johnston, D.; Brager, D.H. Loss of functional A-type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J. Neurosci. 2013, 33, 19442–19450. [Google Scholar] [CrossRef]
- Kalmbach, B.E.; Brager, D.H. Fragile X mental retardation protein modulates somatic D-type K(+) channels and action potential threshold in the mouse prefrontal cortex. J. Neurophysiol. 2020, 124, 1766–1773. [Google Scholar] [CrossRef]
- Selby, L.; Zhang, C.; Sun, Q.Q. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 2007, 412, 227–232. [Google Scholar] [CrossRef]
- D’Hulst, C.; De Geest, N.; Reeve, S.P.; Van Dam, D.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006, 1121, 238–245. [Google Scholar] [CrossRef]
- D’Hulst, C.; Heulens, I.; Brouwer, J.R.; Willemsen, R.; De Geest, N.; Reeve, S.P.; De Deyn, P.P.; Hassan, B.A.; Kooy, R.F. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS). Brain Res. 2009, 1253, 176–183. [Google Scholar] [CrossRef]
- Sabanov, V.; Braat, S.; D’Andrea, L.; Willemsen, R.; Zeidler, S.; Rooms, L.; Bagni, C.; Kooy, R.F.; Balschun, D. Impaired GABAergic inhibition in the hippocampus of Fmr1 knockout mice. Neuropharmacology 2017, 116, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Adusei, D.C.; Pacey, L.K.; Chen, D.; Hampson, D.R. Early developmental alterations in GABAergic protein expression in fragile X knockout mice. Neuropharmacology 2010, 59, 167–171. [Google Scholar] [CrossRef]
- El Idrissi, A.; Ding, X.H.; Scalia, J.; Trenkner, E.; Brown, W.T.; Dobkin, C. Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 2005, 377, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Davidovic, L.; Navratil, V.; Bonaccorso, C.M.; Catania, M.V.; Bardoni, B.; Dumas, M.E. A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res. 2011, 21, 2190–2202. [Google Scholar] [CrossRef] [PubMed]
- Braat, S.; D’Hulst, C.; Heulens, I.; De Rubeis, S.; Mientjes, E.; Nelson, D.L.; Willemsen, R.; Bagni, C.; Van Dam, D.; De Deyn, P.P.; et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle 2015, 14, 2985–2995. [Google Scholar] [CrossRef]
- Wahlstrom-Helgren, S.; Klyachko, V.A. GABAB receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J. Physiol. 2015, 593, 5009–5024. [Google Scholar] [CrossRef]
- Zhang, N.; Peng, Z.; Tong, X.; Lindemeyer, A.K.; Cetina, Y.; Huang, C.S.; Olsen, R.W.; Otis, T.S.; Houser, C.R. Decreased surface expression of the δ subunit of the GABA(A) receptor contributes to reduced tonic inhibition in dentate granule cells in a mouse model of fragile X syndrome. Exp. Neurol. 2017, 297, 168–178. [Google Scholar] [CrossRef]
- Paluszkiewicz, S.M.; Olmos-Serrano, J.L.; Corbin, J.G.; Huntsman, M.M. Impaired inhibitory control of cortical synchronization in fragile X syndrome. J. Neurophysiol. 2011, 106, 2264–2272. [Google Scholar] [CrossRef]
- Conde, V.; Palomar, F.J.; Lama, M.J.; Martínez, R.; Carrillo, F.; Pintado, E.; Mir, P. Abnormal GABA-mediated and cerebellar inhibition in women with the fragile X premutation. J. Neurophysiol. 2013, 109, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Filice, F.; Janickova, L.; Henzi, T.; Bilella, A.; Schwaller, B. The Parvalbumin Hypothesis of Autism Spectrum Disorder. Front. Cell. Neurosci. 2020, 14, 577525. [Google Scholar] [CrossRef]
- Goswami, S.; Cavalier, S.; Sridhar, V.; Huber, K.M.; Gibson, J.R. Local cortical circuit correlates of altered EEG in the mouse model of Fragile X syndrome. Neurobiol. Dis. 2019, 124, 563–572. [Google Scholar] [CrossRef]
- Pedapati, E.V.; Schmitt, L.M.; Ethridge, L.E.; Miyakoshi, M.; Sweeney, J.A.; Liu, R.; Smith, E.; Shaffer, R.C.; Dominick, K.C.; Gilbert, D.L.; et al. Neocortical localization and thalamocortical modulation of neuronal hyperexcitability contribute to Fragile X Syndrome. Commun. Biol. 2022, 5, 442. [Google Scholar] [CrossRef]
- Ethridge, L.E.; White, S.P.; Mosconi, M.W.; Wang, J.; Pedapati, E.V.; Erickson, C.A.; Byerly, M.J.; Sweeney, J.A. Neural synchronization deficits linked to cortical hyper-excitability and auditory hypersensitivity in fragile X syndrome. Mol. Autism 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Pollali, E.; Hollnagel, J.-O.; Çalışkan, G. Hippocampal gamma-band oscillopathy in a mouse model of Fragile X Syndrome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wilson, M.A.; McNaughton, B.L. Reactivation of hippocampal ensemble memories during sleep. Science 1994, 265, 676–679. [Google Scholar]
- Foster, D.J. Replay Comes of Age. Annu. Rev. Neurosci. 2017, 40, 581–602. [Google Scholar] [CrossRef]
- Buzsaki, G. Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 2015, 25, 1073–1188. [Google Scholar] [CrossRef]
- Tomar, A.; Polygalov, D.; Chattarji, S.; McHugh, T.J. Stress enhances hippocampal neuronal synchrony and alters ripple-spike interaction. Neurobiol. Stress 2021, 14, 100327. [Google Scholar] [CrossRef]
- Kuga, N.; Nakayama, R.; Morikawa, S.; Yagishita, H.; Konno, D.; Shiozaki, H.; Honjoya, N.; Ikegaya, Y.; Sasaki, T. Hippocampal sharp wave ripples underlie stress susceptibility in male mice. Nat. Commun. 2023, 14, 2105. [Google Scholar] [CrossRef] [PubMed]
- Papale, A.E.; Zielinski, M.C.; Frank, L.M.; Jadhav, S.P.; Redish, A.D. Interplay between Hippocampal Sharp-Wave-Ripple Events and Vicarious Trial and Error Behaviors in Decision Making. Neuron 2016, 92, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.M.; Shaffer, R.C.; Hessl, D.; Erickson, C. Executive Function in Fragile X Syndrome: A Systematic Review. Brain Sci. 2019, 9, 15. [Google Scholar] [CrossRef]
- Protic, D.D.; Aishworiya, R.; Salcedo-Arellano, M.J.; Tang, S.J.; Milisavljevic, J.; Mitrovic, F.; Hagerman, R.J.; Budimirovic, D.B. Fragile X Syndrome: From Molecular Aspect to Clinical Treatment. Int. J. Mol. Sci. 2022, 23, 1935. [Google Scholar] [CrossRef]
- Cregenzán-Royo, O.; Brun-Gasca, C.; Fornieles-Deu, A. Behavior Problems and Social Competence in Fragile X Syndrome: A Systematic Review. Genes 2022, 13, 280. [Google Scholar] [CrossRef]
- Berzhanskaya, J.; Phillips, M.A.; Shen, J.; Colonnese, M.T. Sensory hypo-excitability in a rat model of fetal development in Fragile X Syndrome. Sci. Rep. 2016, 6, 30769. [Google Scholar] [CrossRef]
- Patel, J.; Lukkes, J.L.; Shekhar, A. Overview of genetic models of autism spectrum disorders. Prog. Brain Res. 2018, 241, 1–36. [Google Scholar] [CrossRef]
- Nomura, T.; Musial, T.F.; Marshall, J.J.; Zhu, Y.; Remmers, C.L.; Xu, J.; Nicholson, D.A.; Contractor, A. Delayed Maturation of Fast-Spiking Interneurons Is Rectified by Activation of the TrkB Receptor in the Mouse Model of Fragile X Syndrome. J. Neurosci. 2017, 37, 11298–11310. [Google Scholar] [CrossRef]
- Goel, A.; Cantu, D.A.; Guilfoyle, J.; Chaudhari, G.R.; Newadkar, A.; Todisco, B.; de Alba, D.; Kourdougli, N.; Schmitt, L.M.; Pedapati, E.; et al. Impaired perceptual learning in a mouse model of Fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat. Neurosci. 2018, 21, 1404–1411. [Google Scholar] [CrossRef]
- Wen, T.H.; Afroz, S.; Reinhard, S.M.; Palacios, A.R.; Tapia, K.; Binder, D.K.; Razak, K.A.; Ethell, I.M. Genetic Reduction of Matrix Metalloproteinase-9 Promotes Formation of Perineuronal Nets Around Parvalbumin-Expressing Interneurons and Normalizes Auditory Cortex Responses in Developing Fmr1 Knock-Out Mice. Cereb. Cortex 2018, 28, 3951–3964. [Google Scholar] [CrossRef]
- Wen, T.H.; Binder, D.K.; Ethell, I.M.; Razak, K.A. The Perineuronal ‘Safety’ Net? Perineuronal Net Abnormalities in Neurological Disorders. Front. Mol. Neurosci. 2018, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.H.F.; Lai, T.K.Y.; Su, P.; Liu, F. Altered cortical Cytoarchitecture in the Fmr1 knockout mouse. Mol. Brain 2019, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Reyes, S.T.; Mohajeri, S.; Krasinska, K.; Guo, S.G.; Gu, M.; Pisani, L.; Rosenberg, J.; Spielman, D.M.; Chin, F.T. GABA Measurement in a Neonatal Fragile X Syndrome Mouse Model Using (1)H-Magnetic Resonance Spectroscopy and Mass Spectrometry. Front. Mol. Neurosci. 2020, 13, 612685. [Google Scholar] [CrossRef]
- Pouchelon, G.; Dwivedi, D.; Bollmann, Y.; Agba, C.K.; Xu, Q.; Mirow, A.M.C.; Kim, S.; Qiu, Y.; Sevier, E.; Ritola, K.D.; et al. The organization and development of cortical interneuron presynaptic circuits are area specific. Cell Rep. 2021, 37, 109993. [Google Scholar] [CrossRef]
- Castagnola, S.; Cazareth, J.; Lebrigand, K.; Jarjat, M.; Magnone, V.; Delhaye, S.; Brau, F.; Bardoni, B.; Maurin, T. Agonist-induced functional analysis and cell sorting associated with single-cell transcriptomics characterizes cell subtypes in normal and pathological brain. Genome Res. 2020, 30, 1633–1642. [Google Scholar] [CrossRef]
- Rais, M.; Lovelace, J.W.; Shuai, X.S.; Woodard, W.; Bishay, S.; Estrada, L.; Sharma, A.R.; Nguy, A.; Kulinich, A.; Pirbhoy, P.S.; et al. Functional consequences of postnatal interventions in a mouse model of Fragile X syndrome. Neurobiol. Dis. 2022, 162, 105577. [Google Scholar] [CrossRef]
- He, Q.; Arroyo, E.D.; Smukowski, S.N.; Xu, J.; Piochon, C.; Savas, J.N.; Portera-Cailliau, C.; Contractor, A. Critical period inhibition of NKCC1 rectifies synapse plasticity in the somatosensory cortex and restores adult tactile response maps in fragile X mice. Mol. Psychiatry 2019, 24, 1732–1747. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, M.; van der Lei, M.B.; Kitiashvili, M.; Mamcarz, M.; Oliveira, M.M.; Longo, F.; Klann, E. Deletion of Fmr1 in parvalbumin-expressing neurons results in dysregulated translation and selective behavioral deficits associated with fragile X syndrome. Mol. Autism 2022, 13, 29. [Google Scholar] [CrossRef]
- Olmos-Serrano, J.L.; Paluszkiewicz, S.M.; Martin, B.S.; Kaufmann, W.E.; Corbin, J.G.; Huntsman, M.M. Defective GABAergic neurotransmission and pharmacological rescue of neuronal hyperexcitability in the amygdala in a mouse model of fragile X syndrome. J. Neurosci. 2010, 30, 9929–9938. [Google Scholar] [CrossRef]
- Vislay, R.L.; Martin, B.S.; Olmos-Serrano, J.L.; Kratovac, S.; Nelson, D.L.; Corbin, J.G.; Huntsman, M.M. Homeostatic responses fail to correct defective amygdala inhibitory circuit maturation in fragile X syndrome. J. Neurosci. 2013, 33, 7548–7558. [Google Scholar] [CrossRef]
- Lovelace, J.W.; Rais, M.; Palacios, A.R.; Shuai, X.S.; Bishay, S.; Popa, O.; Pirbhoy, P.S.; Binder, D.K.; Nelson, D.L.; Ethell, I.M.; et al. Deletion of Fmr1 from Forebrain Excitatory Neurons Triggers Abnormal Cellular, EEG, and Behavioral Phenotypes in the Auditory Cortex of a Mouse Model of Fragile X Syndrome. Cereb. Cortex 2020, 30, 969–988. [Google Scholar] [CrossRef]
- Martin, B.S.; Corbin, J.G.; Huntsman, M.M. Deficient tonic GABAergic conductance and synaptic balance in the fragile X syndrome amygdala. J. Neurophysiol. 2014, 112, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Arsenault, J.; Bah, A.; Krzeminski, M.; Fekete, A.; Chao, O.Y.; Pacey, L.K.; Wang, A.; Forman-Kay, J.; Hampson, D.R.; et al. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Mol. Psychiatry 2020, 25, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Cea-Del Rio, C.A.; Nunez-Parra, A.; Freedman, S.M.; Kushner, J.K.; Alexander, A.L.; Restrepo, D.; Huntsman, M.M. Disrupted inhibitory plasticity and homeostasis in Fragile X syndrome. Neurobiol. Dis. 2020, 142, 104959. [Google Scholar] [CrossRef]
- Cellot, G.; Cherubini, E. Reduced inhibitory gate in the barrel cortex of Neuroligin3R451C knock-in mice, an animal model of autism spectrum disorders. Physiol. Rep. 2014, 2, e12077. [Google Scholar] [CrossRef]
- Curia, G.; Papouin, T.; Séguéla, P.; Avoli, M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb. Cortex 2009, 19, 1515–1520. [Google Scholar] [CrossRef]
- Howard, M.A.; Rubenstein, J.L.; Baraban, S.C. Bidirectional homeostatic plasticity induced by interneuron cell death and transplantation in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 492–497. [Google Scholar] [CrossRef]
- Asiminas, A.; Booker, S.A.; Dando, O.R.; Kozic, Z.; Arkell, D.; Inkpen, F.H.; Sumera, A.; Akyel, I.; Kind, P.C.; Wood, E.R. Experience-dependent changes in hippocampal spatial activity and hippocampal circuit function are disrupted in a rat model of Fragile X Syndrome. Mol. Autism 2022, 13, 49. [Google Scholar] [CrossRef]
- Deng, P.Y.; Kumar, A.; Cavalli, V.; Klyachko, V.A. FMRP regulates GABA(A) receptor channel activity to control signal integration in hippocampal granule cells. Cell Rep. 2022, 39, 110820. [Google Scholar] [CrossRef]
- Cellot, G.; Maggi, L.; Di Castro, M.A.; Catalano, M.; Migliore, R.; Migliore, M.; Scattoni, M.L.; Calamandrei, G.; Cherubini, E. Premature changes in neuronal excitability account for hippocampal network impairment and autistic-like behavior in neonatal BTBR T+tf/J mice. Sci. Rep. 2016, 6, 31696. [Google Scholar] [CrossRef]
- Hong, A.; Zhang, A.; Ke, Y.; El Idrissi, A.; Shen, C.H. Downregulation of GABA(A) β subunits is transcriptionally controlled by Fmr1p. J. Mol. Neurosci. MN 2012, 46, 272–275. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Monday, H.R.; Yan, J.; Gompers, A.; Buxbaum, A.R.; Sawicka, K.J.; Singer, R.H.; Castillo, P.E.; Zukin, R.S. CPEB3-dependent increase in GluA2 subunits impairs excitatory transmission onto inhibitory interneurons in a mouse model of fragile X. Cell Rep. 2022, 39, 110853. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Chadchankar, J.; Vien, T.N.; Mighdoll, M.I.; Hyde, T.M.; Mather, R.J.; Deeb, T.Z.; Pangalos, M.N.; Brandon, N.J.; Dunlop, J.; et al. Deficits in the activity of presynaptic γ-aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J. Biol. Chem. 2017, 292, 6621–6632. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, J.; Violante, I.R.; Sereno, J.; Leitão, R.A.; Cai, Y.; Abrunhosa, A.; Silva, A.P.; Silva, A.J.; Castelo-Branco, M. Testing the excitation/inhibition imbalance hypothesis in a mouse model of the autism spectrum disorder: In vivo neurospectroscopy and molecular evidence for regional phenotypes. Mol. Autism 2017, 8, 47. [Google Scholar] [CrossRef]
- Eichenbaum, H. Memory: Organization and Control. Annu. Rev. Psychol. 2017, 68, 19–45. [Google Scholar] [CrossRef]
- Goode, T.D.; Tanaka, K.Z.; Sahay, A.; McHugh, T.J. An Integrated Index: Engrams, Place Cells, and Hippocampal Memory. Neuron 2020, 107, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Moscovitch, M.; Rosenbaum, R.S.; Gilboa, A.; Addis, D.R.; Westmacott, R.; Grady, C.; McAndrews, M.P.; Levine, B.; Black, S.; Winocur, G.; et al. Functional neuroanatomy of remote episodic, semantic and spatial memory: A unified account based on multiple trace theory. J. Anat. 2005, 207, 35–66. [Google Scholar] [CrossRef]
- Pronier, É.; Morici, J.F.; Girardeau, G. The role of the hippocampus in the consolidation of emotional memories during sleep. Trends Neurosci. 2023, 46, 912–925. [Google Scholar] [CrossRef]
- Amaral, D.G.; Lavenex, P. Hippocampal Neuroanatomy. In The Hippocampus Book; Andersen, P., Morris, R., Amaral, D., Bliss, T., O’Keefe, J., Eds.; Oxford University Press: Oxford, UK, 2007; pp. 37–114. [Google Scholar]
- Pelkey, K.A.; Chittajallu, R.; Craig, M.T.; Tricoire, L.; Wester, J.C.; McBain, C.J. Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev. 2017, 97, 1619–1747. [Google Scholar] [CrossRef]
- Caroni, P. Inhibitory microcircuit modules in hippocampal learning. Curr. Opin. Neurobiol. 2015, 35, 66–73. [Google Scholar] [CrossRef]
- Small, S.A. The longitudinal axis of the hippocampal formation: Its anatomy, circuitry, and role in cognitive function. Rev. Neurosci. 2002, 13, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bast, T. The hippocampal learning-behavior translation and the functional significance of hippocampal dysfunction in schizophrenia. Curr. Opin. Neurobiol. 2011, 21, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Risold, P.Y.; Swanson, L.W. Structural evidence for functional domains in the rat hippocampus. Science 1996, 272, 1484–1486. [Google Scholar]
- Poppenk, J.; Evensmoen, H.R.; Moscovitch, M.; Nadel, L. Long-axis specialization of the human hippocampus. Trends Cogn. Sci. 2013, 17, 230–240. [Google Scholar] [CrossRef]
- Cembrowski, M.S.; Spruston, N. Heterogeneity within classical cell types is the rule: Lessons from hippocampal pyramidal neurons. Nat. Rev. Neurosci. 2019, 20, 193–204. [Google Scholar] [CrossRef]
- van Strien, N.M.; Cappaert, N.L.; Witter, M.P. The anatomy of memory: An interactive overview of the parahippocampal-hippocampal network. Nat. Rev. Neurosci. 2009, 10, 272–282. [Google Scholar] [CrossRef]
- Jin, J.; Maren, S. Prefrontal-Hippocampal Interactions in Memory and Emotion. Front. Syst. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef]
- Morris, R.G.; Garrud, P.; Rawlins, J.N.; O’Keefe, J. Place navigation impaired in rats with hippocampal lesions. Nature 1982, 297, 681–683. [Google Scholar] [CrossRef]
- Maguire, E.A. Hippocampal involvement in human topographical memory: Evidence from functional imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 1475–1480. [Google Scholar] [CrossRef]
- Moscovitch, M.; Nadel, L.; Winocur, G.; Gilboa, A.; Rosenbaum, R.S. The cognitive neuroscience of remote episodic, semantic and spatial memory. Curr. Opin. Neurobiol. 2006, 16, 179–190. [Google Scholar] [CrossRef]
- Eichenbaum, H.; Cohen, N.J. Can we reconcile the declarative memory and spatial navigation views on hippocampal function? Neuron 2014, 83, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; Ostrander, M.M.; Mueller, N.K.; Figueiredo, H. Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adrenocortical axis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 1201–1213. [Google Scholar]
- Preston, A.R.; Eichenbaum, H. Interplay of hippocampus and prefrontal cortex in memory. Curr. Biol. CB 2013, 23, R764–R773. [Google Scholar] [CrossRef]
- Montagrin, A.; Saiote, C.; Schiller, D. The social hippocampus. Hippocampus 2018, 28, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Keppler, E.; Molas, S. Prompting social investigation. eLife 2024, 13, e99363. [Google Scholar] [CrossRef]
- Hagerman, P.J.; Stafstrom, C.E. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef]
- Qiu, L.F.; Lu, T.J.; Hu, X.L.; Yi, Y.H.; Liao, W.P.; Xiong, Z.Q. Limbic epileptogenesis in a mouse model of fragile X syndrome. Cereb. Cortex 2009, 19, 1504–1514. [Google Scholar] [CrossRef]
- Devine, I.M.; Stafstrom, C.E. Fragile X syndrome. In The Causes of Epilepsy: Common and Uncommon Causes in Adults and Children; Shorvon, S.D., Andermann, F., Guerrini, R., Eds.; Cambridge University Press: Cambridge, UK, 2011; pp. 272–276. [Google Scholar]
- Hall, S.S.; Jiang, H.; Reiss, A.L.; Greicius, M.D. Identifying large-scale brain networks in fragile X syndrome. JAMA Psychiatry 2013, 70, 1215–1223. [Google Scholar] [CrossRef]
- Arbab, T.; Battaglia, F.P.; Pennartz, C.M.A.; Bosman, C.A. Abnormal hippocampal theta and gamma hypersynchrony produces network and spike timing disturbances in the Fmr1-KO mouse model of Fragile X syndrome. Neurobiol. Dis. 2018, 114, 65–73. [Google Scholar] [CrossRef]
- Chrobak, J.J.; Buzsáki, G. Selective activation of deep layer (V-VI) retrohippocampal cortical neurons during hippocampal sharp waves in the behaving rat. J. Neurosci. 1994, 14, 6160–6170. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Villegas, J.F.; Willeke, K.F.; Logothetis, N.K.; Besserve, M. Dissecting the Synapse- and Frequency-Dependent Network Mechanisms of In Vivo Hippocampal Sharp Wave-Ripples. Neuron 2018, 100, 1224–1240.e1213. [Google Scholar] [CrossRef] [PubMed]
- Klausberger, T.; Somogyi, P. Neuronal diversity and temporal dynamics: The unity of hippocampal circuit operations. Science 2008, 321, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Schlingloff, D.; Kali, S.; Freund, T.F.; Hajos, N.; Gulyas, A.I. Mechanisms of sharp wave initiation and ripple generation. J. Neurosci. 2014, 34, 11385–11398. [Google Scholar] [CrossRef]
- Melonakos, E.D.; White, J.A.; Fernandez, F.R. A model of cholinergic suppression of hippocampal ripples through disruption of balanced excitation/inhibition. Hippocampus 2019, 29, 773–786. [Google Scholar] [CrossRef]
- Ecker, A.; Bagi, B.; Vértes, E.; Steinbach-Németh, O.; Karlócai, M.R.; Papp, O.I.; Miklós, I.; Hájos, N.; Freund, T.F.; Gulyás, A.I.; et al. Hippocampal sharp wave-ripples and the associated sequence replay emerge from structured synaptic interactions in a network model of area CA3. eLife 2022, 11, e71850. [Google Scholar] [CrossRef]
- Trompoukis, G.; Rigas, P.; Leontiadis, L.J.; Papatheodoropoulos, C. Ih, GIRK, and KCNQ/Kv7 channels differently modulate sharp wave-ripples in the dorsal and ventral hippocampus. Mol. Cell. Neurosci. 2020, 107, 103531. [Google Scholar] [CrossRef]
- Contreras, A.; Djebari, S.; Temprano-Carazo, S.; Múnera, A.; Gruart, A.; Delgado-Garcia, J.M.; Jiménez-Díaz, L.; Navarro-López, J.D. Impairments in hippocampal oscillations accompany the loss of LTP induced by GIRK activity blockade. Neuropharmacology 2023, 238, 109668. [Google Scholar] [CrossRef]
- Richter, J.P.; Behrens, C.J.; Chakrabarty, A.; Heinemann, U. Effects of 4-aminopyridine on sharp wave-ripples in rat hippocampal slices. Neuroreport 2008, 19, 491–496. [Google Scholar] [CrossRef]
- Simeone, T.A.; Simeone, K.A.; Samson, K.K.; Kim, D.Y.; Rho, J.M. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol. Dis. 2013, 54, 68–81. [Google Scholar] [CrossRef]
- Papatheodoropoulos, C.; Sotiriou, E.; Kotzadimitriou, D.; Drimala, P. At clinically relevant concentrations the anaesthetic/amnesic thiopental but not the anticonvulsant phenobarbital interferes with hippocampal sharp wave-ripple complexes. BMC Neurosci. 2007, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoropoulos, C. Patterned activation of hippocampal network (approximately 10 Hz) during in vitro sharp wave-ripples. Neuroscience 2010, 168, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, P.; Papatheodoropoulos, C. Effects of μ-opioid receptor modulation on the hippocampal network activity of sharp wave and ripples. Br. J. Pharmacol. 2013, 168, 1146–1164. [Google Scholar] [CrossRef] [PubMed]
- Pochinok, I.; Stöber, T.M.; Triesch, J.; Chini, M.; Hanganu-Opatz, I.L. A developmental increase of inhibition promotes the emergence of hippocampal ripples. Nat. Commun. 2024, 15, 738. [Google Scholar] [CrossRef]
- Caliskan, G.; Stork, O. Hippocampal network oscillations at the interplay between innate anxiety and learned fear. Psychopharmacology 2019, 236, 321–338. [Google Scholar] [CrossRef]
- Anagnostou, E.; Taylor, M.J. Review of neuroimaging in autism spectrum disorders: What have we learned and where we go from here. Mol. Autism 2011, 2, 4. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Fetit, R.; Hillary, R.F.; Price, D.J.; Lawrie, S.M. The neuropathology of autism: A systematic review of post-mortem studies of autism and related disorders. Neurosci. Biobehav. Rev. 2021, 129, 35–62. [Google Scholar] [CrossRef]
- Razak, K.A.; Dominick, K.C.; Erickson, C.A. Developmental studies in fragile X syndrome. J. Neurodev. Disord. 2020, 12, 13. [Google Scholar] [CrossRef]

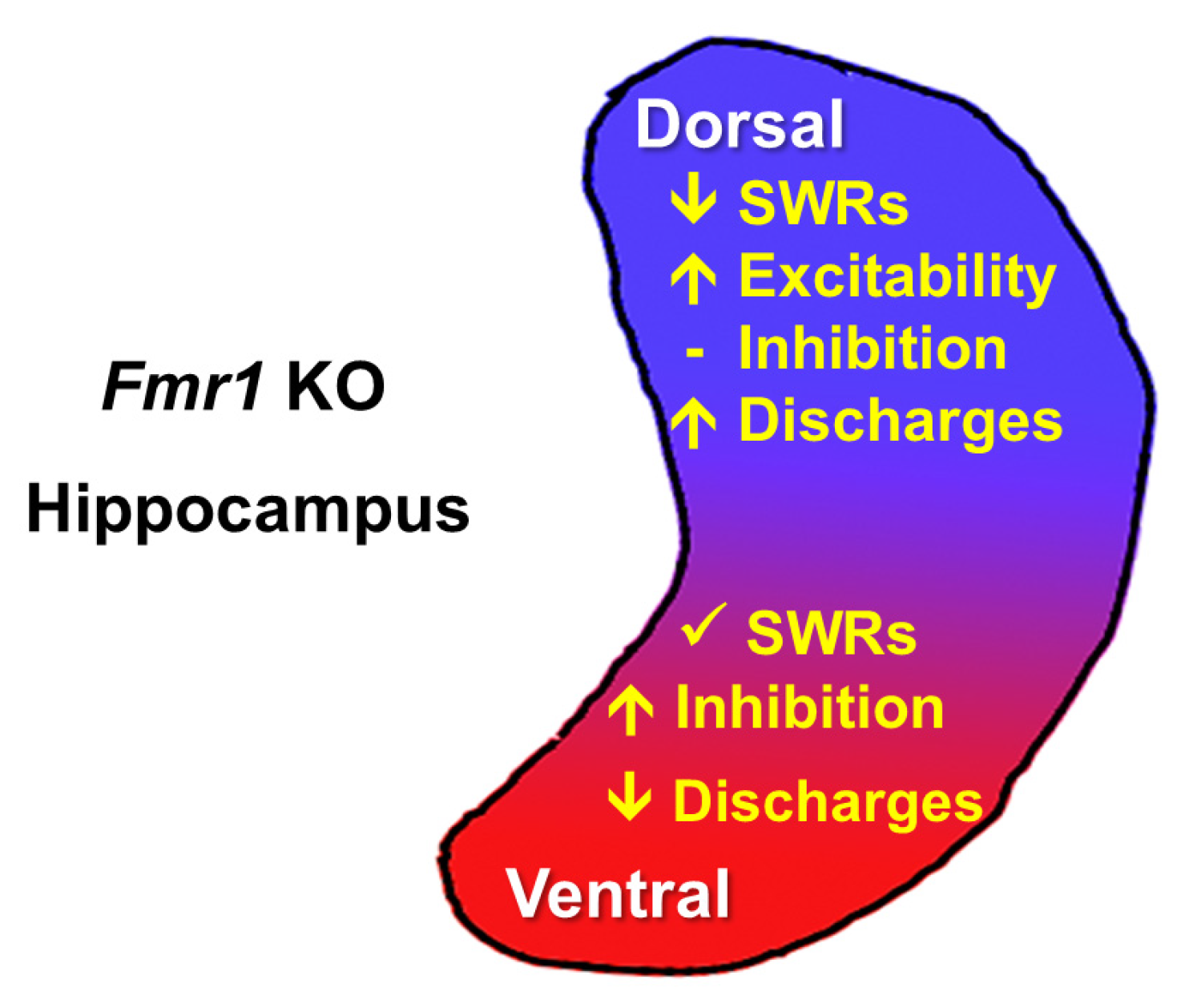
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papatheodoropoulos, C. Compensatory Regulation of Excitation/Inhibition Balance in the Ventral Hippocampus: Insights from Fragile X Syndrome. Biology 2025, 14, 363. https://doi.org/10.3390/biology14040363
Papatheodoropoulos C. Compensatory Regulation of Excitation/Inhibition Balance in the Ventral Hippocampus: Insights from Fragile X Syndrome. Biology. 2025; 14(4):363. https://doi.org/10.3390/biology14040363
Chicago/Turabian StylePapatheodoropoulos, Costas. 2025. "Compensatory Regulation of Excitation/Inhibition Balance in the Ventral Hippocampus: Insights from Fragile X Syndrome" Biology 14, no. 4: 363. https://doi.org/10.3390/biology14040363
APA StylePapatheodoropoulos, C. (2025). Compensatory Regulation of Excitation/Inhibition Balance in the Ventral Hippocampus: Insights from Fragile X Syndrome. Biology, 14(4), 363. https://doi.org/10.3390/biology14040363