
Visualization of Runs of Homozygosity and Classification Using Convolutional Neural Networks
 , , , , , , , , and
, , , , , , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Traits
2.2. Genotyping and Quality Control
2.3. Analysis of Homozygosity (ROH)
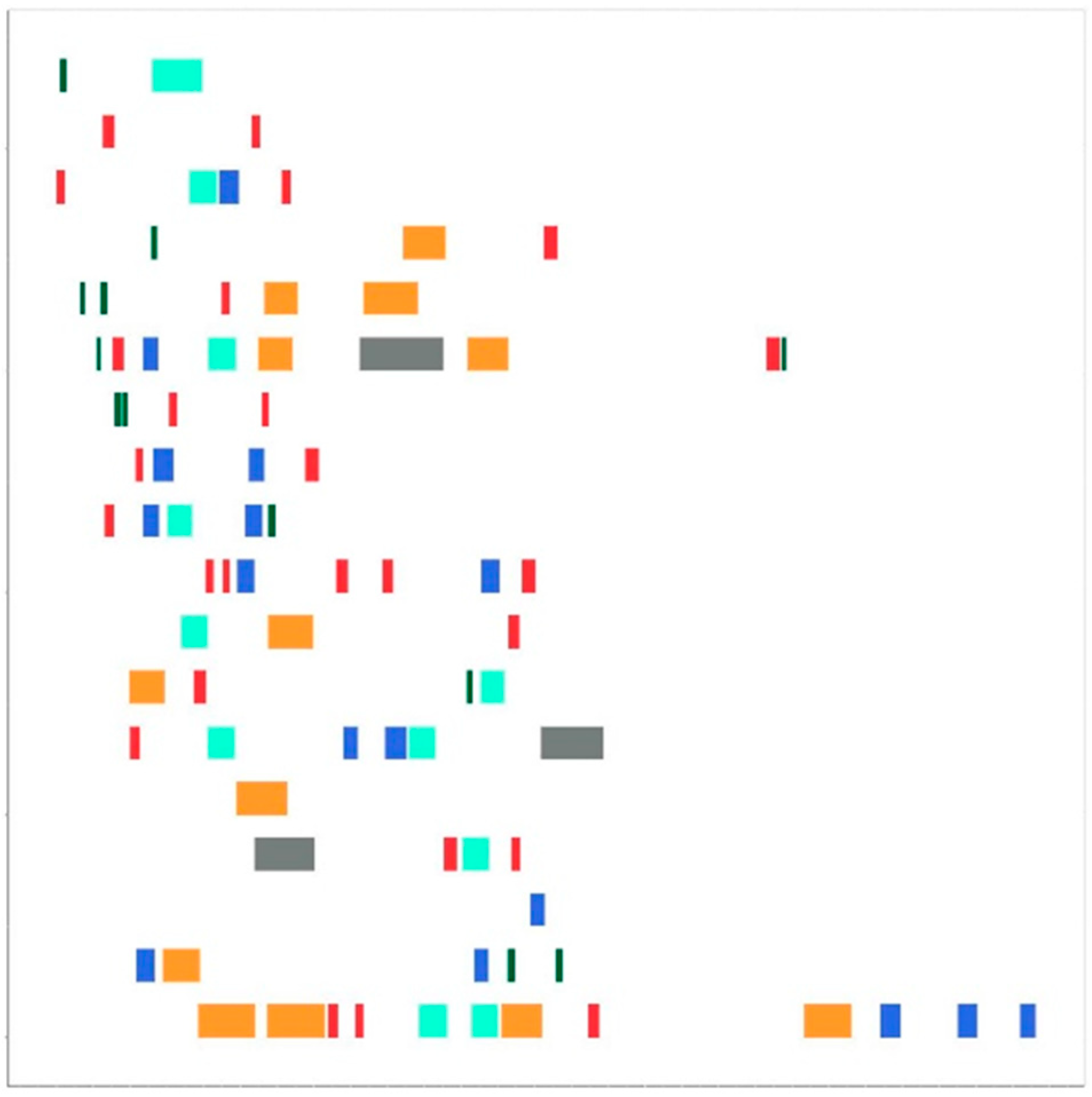
2.4. Drawing Maps
2.5. Convolutional Neural Network (CNN) Model
2.6. Cross-Validation and Comparison with Classical Methods
2.7. Identifying Informative Regions
3. Results
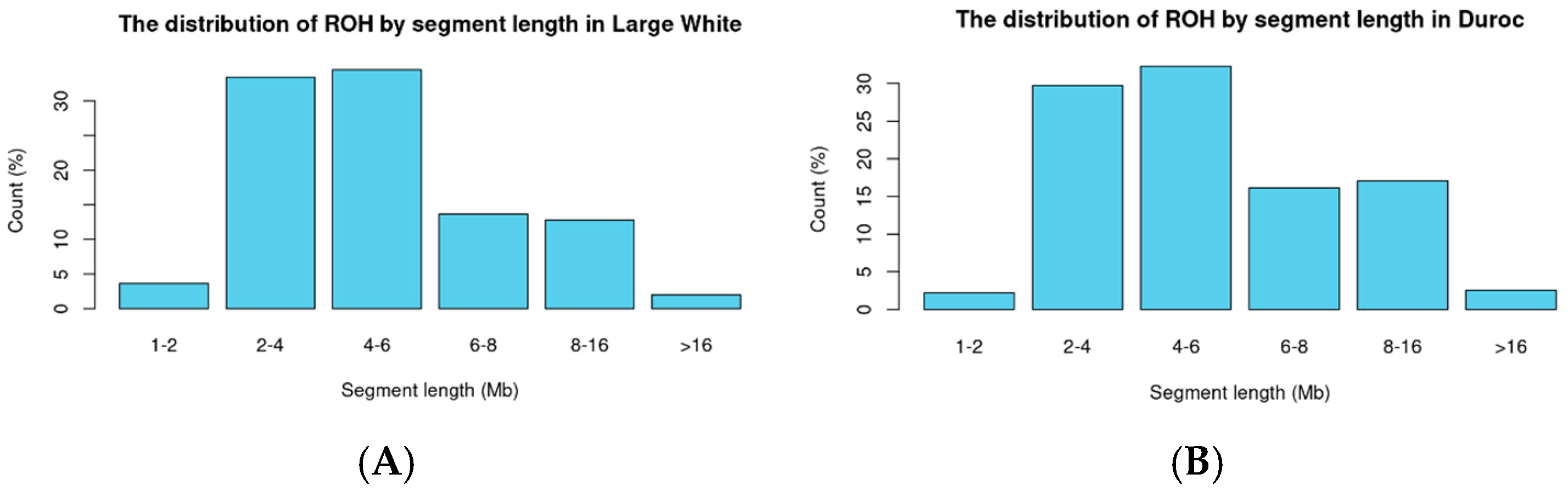
3.1. Classification by Breed in Pigs
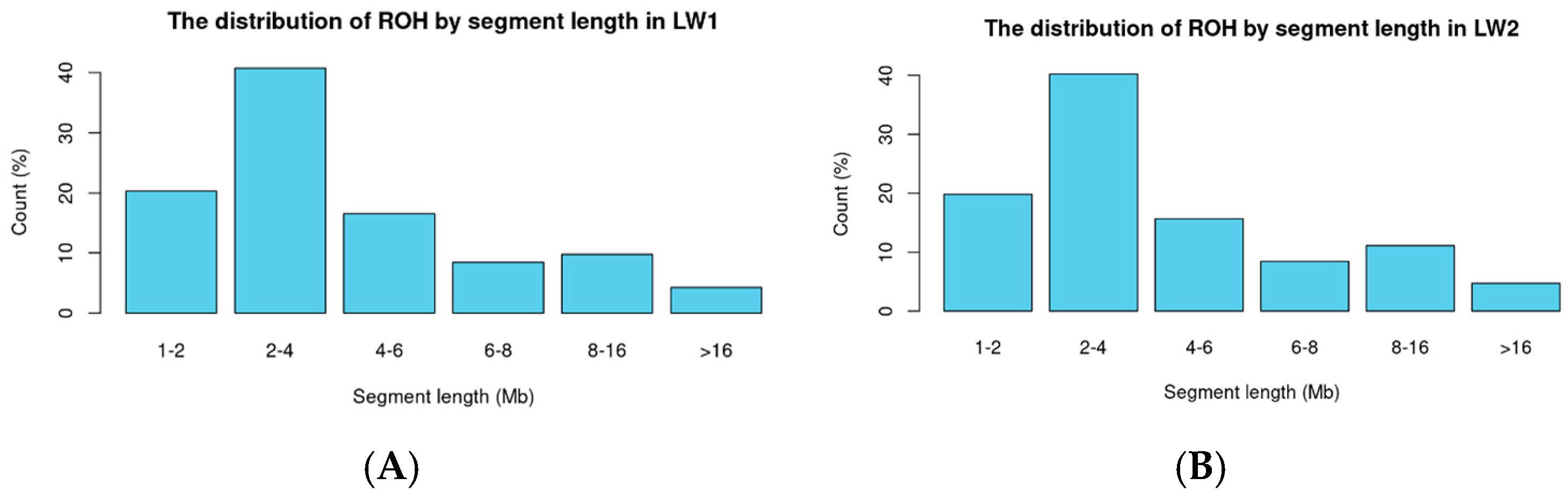
3.2. Binary Trait Classification in Large White Pigs
3.3. Ensuring Effective Model Operation
4. Discussion
Limitations and Prospects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Fieder, M.; Mitchell, B.L.; Gordon, S.; Huber, S.; Martin, N.G. Ethnic Identity and Genome Wide Runs of Homozygosity. Behav. Genet. 2021, 51, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.P.; Silva, M.V.G.B.; Lima, A.L.F.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Falchi, L.; Cesarani, A.; Mastrangelo, S.; Senczuk, G.; Portolano, B.; Pilla, F.; Macciotta, N.P.P. Analysis of runs of homozygosity of cattle living in different climate zones. J. Anim. Sci. 2023, 101, skad061. [Google Scholar] [CrossRef] [PubMed]
- Lemes, R.B.; Nunes, K.; Carnavalli, J.E.P.; Kimura, L.; Mingroni-Netto, R.C.; Meyer, D.; Otto, P.A. Inbreeding estimates in human populations: Applying new approaches to an admixed Brazilian isolate. PLoS ONE 2018, 13, e0196360. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Mak, A.C.Y.; White, M.J.; Hu, D.; Eng, C.; Burchard, E.G.; Hernandez, R.D. Ancestry-Dependent Enrichment of Deleterious Homozygotes in Runs of Homozygosity. Am. J. Hum. Genet. 2019, 105, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Ehlers, C.L. Long tracks of homozygosity predict the severity of alcohol use disorders in an American Indian population. Mol. Psychiatry 2021, 26, 2200–2211. [Google Scholar] [CrossRef]
- Clark, D.W.; Okada, Y.; Moore, K.H.; Mason, D.; Pirastu, N.; Gandin, I.; Mattsson, H.; Barnes, C.L.K.; Lin, K.; Zhao, J.H.; et al. Associations of autozygosity with a broad range of human phenotypes. Nat. Commun. 2019, 10, 4957. [Google Scholar] [CrossRef]
- Ojeda-Marín, C.; Gutiérrez, J.P.; Formoso-Rafferty, N.; Goyache, F.; Cervantes, I. Differential patterns in runs of homozygosity in two mice lines under divergent selection for environmental variability for birth weight. J. Anim. Breed. Genet. 2024, 141, 193–206. [Google Scholar] [CrossRef]
- Becker, G.M.; Thorne, J.W.; Burke, J.M.; Lewis, R.M.; Notter, D.R.; Morgan, J.L.; Schauer, C.S.; Stewart, W.C.; Redden, R.R.; Murdoch, B.M. Genetic diversity of United States Rambouillet, Katahdin and Dorper sheep. Genet. Sel. Evol. 2024, 56, 56. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Dall’Olio, S.; Costa, L.N.; Tinarelli, S.; Gallo, M.; Fontanesi, L. Runs of homozygosity islands in Italian cosmopolitan and autochthonous pig breeds identify selection signatures in the porcine genome. Livest. Sci. 2020, 240, 104219. [Google Scholar] [CrossRef]
- Di Gregorio, P.; Perna, A.; Di Trana, A.; Rando, A. Identification of ROH Islands Conserved through Generations in Pigs Belonging to the Nero Lucano Breed. Genes 2023, 14, 1503. [Google Scholar] [CrossRef]
- Bakoev, S.; Kolosov, A.; Bakoev, F.; Kostyunina, O.; Bakoev, N.; Romanets, T.; Koshkina, O.; Getmantseva, L. Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia. Life 2021, 11, 861. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Ribani, A.; Moscatelli, G.; Bonacini, M.; Prandi, M.; Mancin, E.; Mantovani, R.; Dall’Olio, S.; Fontanesi, L. Comparative analysis of inbreeding parameters and runs of homozygosity islands in 2 Italian autochthonous cattle breeds mainly raised in the Parmigiano-Reggiano cheese production region. J. Dairy science 2022, 105, 2408–2425. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Liu, J.; Zhang, W.; Zhou, M.; Wang, J.; Liu, L.; Su, S.; Zhao, F.; Chen, H.; et al. Genome-wide detection of genetic structure and runs of homozygosity analysis in Anhui indigenous and Western commercial pig breeds using PorcineSNP80k data. BMC Genom. 2022, 23, 373. [Google Scholar] [CrossRef]
- Bakoev, S.Y.; Romanets, T.S.; Korobeynikova, A.V.; Mishina, A.I.; Kolosova, M.A.; Romanets, E.A.; Kolosov, A.Y.; Getmantseva, L.V. Genetic Diversity and Autozygosity Estimates in Wild European Boars and Domesticated Pigs. Ecol. Genet. 2023, 21, 343–355. [Google Scholar] [CrossRef]
- Toratani, M.; Konno, M.; Asai, A.; Koseki, J.; Kawamoto, K.; Tamari, K.; Li, Z.; Sakai, D.; Kudo, T.; Satoh, T.; et al. A convolutional neural network uses microscopic images to differentiate between mouse and human cell lines and their radioresistant clones. Cancer Res. 2018, 78, 6703–6707. [Google Scholar] [CrossRef]
- Shankara, C.; Hariprasad, S.A.; Latha, D.U. Detection of Lung Cancer Using Convolution Neural Network. SN Comput. Sci. 2023, 4, 225. [Google Scholar] [CrossRef]
- Xie, Y.; Zaccagna, F.; Rundo, L.; Testa, C.; Agati, R.; Lodi, R.; Manners, D.N.; Tonon, C. Convolutional neural network techniques for brain tumor classification (from 2015 to 2022): Review, challenges, and future perspectives. Diagnostics 2022, 12, 1850. [Google Scholar] [CrossRef]
- Vu, P.T.; Chahine, C.; Chatterjee, N.; MacLean, M.T.; Swago, S.; Bhattaru, A.; Thompson, E.W.; Ikhlas, A.; Oteng, E.; Davidson, L.; et al. CT imaging-derived phenotypes for abdominal muscle and their association with age and sex in a medical biobank. Sci. Rep. 2024, 14, 14807. [Google Scholar] [CrossRef]
- Kamilaris, A.; Prenafeta-Boldú, F.X. A review of the use of convolutional neural networks in agriculture. J. Agric. Sci. 2018, 156, 312–322. [Google Scholar] [CrossRef]
- Umurungi, S.N.; Ufitikirezi, J.D.D.M.; Zoubek, T.; Bumbálek, R.; Kuneš, R.; Šramhauser, K.; Stehlík, R.; Havelka, Z.; Olšan, P.; Bartoš, P. Leveraging the potential of convolutional neural networks in poultry farming: A 5-year overview. World’s Poult. Sci. J. 2025, 1, 3–34. [Google Scholar] [CrossRef]
- Barber, F.B.N.; Oueslati, A.E. Human exons and introns classification using pre-trained Resnet-50 and GoogleNet models and 13-layers CNN model. J. Genet. Eng. Biotechnol. 2024, 22, 100359. [Google Scholar]
- Chen, X.; Chen, D.G.; Zhao, Z.; Zhan, J.; Ji, C.; Chen, J. Artificial image objects for classification of schizophrenia with GWAS-selected SNVs and convolutional neural network. Patterns 2021, 2, 100303. [Google Scholar] [CrossRef]
- Kupperman, M.D.; Leitner, T.; Ke, R. A deep learning approach to real-time HIV outbreak detection using genetic data. PLoS Comput. Biol. 2022, 18, e1010598. [Google Scholar] [CrossRef]
- Mahin, K.F.; Robiuddin, M.; Islam, M.; Ashraf, S.; Yeasmin, F.; Shatabda, S. PanClassif: Improving pan cancer classification of single cell RNA-seq gene expression data using machine learning. Genomics 2022, 114, 110264. [Google Scholar] [CrossRef]
- Bakoev, S.; Getmantseva, L.; Kolosova, M.; Bakoev, F.; Kolosov, A.; Romanets, E.; Shevtsova, V.; Romanets, T.; Kolosov, Y.; Usatov, A. Identifying Significant SNPs of the Total Number of Piglets Born and Their Relationship with Leg Bumps in Pigs. Biology 2024, 13, 1034. [Google Scholar] [CrossRef]
- Getmantseva, L.; Kolosova, M.; Fede, K.; Korobeinikova, A.; Kolosov, A.; Romanets, E.; Bakoev, F.; Romanets, T.; Yudin, V.; Keskinov, A.; et al. Finding predictors of leg defects in pigs using CNV-GWAS. Genes 2023, 14, 2054. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Figshare. Available online: https://figshare.com/articles/dataset/Chip_data_for_a_Duroc_pig_population/7885703/2?file=14677688 (accessed on 21 February 2025).
- Zhou, J.; Liu, L.; Lopdell, T.J.; Garrick, D.J.; Shi, Y. HandyCNV: Standardized summary, annotation, comparison, and visualization of copy number variant, copy number variation region, and runs of homozygosity. Front. Genet. 2021, 12, 731355. [Google Scholar] [CrossRef]
- Kadir, T.; Brady, M. Saliency, scale and image description. Int. J. Comput. Vis. 2001, 45, 83–105. [Google Scholar] [CrossRef]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep Inside Convolutional Networks: Visualising Image Classification Models and Saliency Maps. arXiv 2013, arXiv:1312.6034. [Google Scholar]
- Gonzalez, R.C.; Woods, R.E. Digital Image Processing, 4th ed.; Pearson Education: New York, NY, USA, 2018. [Google Scholar]
- Druet, T.; Gautier, M. A model-based approach to characterize individual inbreeding at both global and local genomic scales. Mol. Ecol. 2017, 26, 5820–5841. [Google Scholar] [CrossRef]
- Bakoev, S.Y.; Getmantseva, L.V. Methods for assessing inbreeding and selection signatures of farm animals based on extended homozygous regions. Achiev. Sci. Technol. AIC 2019, 33, 63–68. [Google Scholar]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Observed Values | ||||
|---|---|---|---|---|
| Predicted Values | LW | D | ||
| LW | 30 | 0 | ||
| D | 0 | 37 | ||
| Accuracy | Sensitivity | Specificity | Positive Predictive Value | Negative Predictive Value |
| 1.0 (95% CI (0.95)) | 1.0 | 1.0 | 1.0 | 1.0 |
| Observed Values | ||||
|---|---|---|---|---|
| Predicted Values | LW1 | LW2 | ||
| LW1 | 22 | 4 | ||
| LW2 | 5 | 11 | ||
| Accuracy | Sensitivity | Specificity | Positive Predictive Value | Negative Predictive Value |
| 0.7857 95% CI: (0.6319, 0.897) | 0.7333 | 0.8148 | 0.6875 | 0.8462 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakoev, S.; Kolosova, M.; Romanets, T.; Bakoev, F.; Kolosov, A.; Romanets, E.; Korobeinikova, A.; Bakoeva, I.; Akhmedli, V.; Getmantseva, L. Visualization of Runs of Homozygosity and Classification Using Convolutional Neural Networks. Biology 2025, 14, 426. https://doi.org/10.3390/biology14040426
Bakoev S, Kolosova M, Romanets T, Bakoev F, Kolosov A, Romanets E, Korobeinikova A, Bakoeva I, Akhmedli V, Getmantseva L. Visualization of Runs of Homozygosity and Classification Using Convolutional Neural Networks. Biology. 2025; 14(4):426. https://doi.org/10.3390/biology14040426
Chicago/Turabian StyleBakoev, Siroj, Maria Kolosova, Timofey Romanets, Faridun Bakoev, Anatoly Kolosov, Elena Romanets, Anna Korobeinikova, Ilona Bakoeva, Vagif Akhmedli, and Lyubov Getmantseva. 2025. "Visualization of Runs of Homozygosity and Classification Using Convolutional Neural Networks" Biology 14, no. 4: 426. https://doi.org/10.3390/biology14040426
APA StyleBakoev, S., Kolosova, M., Romanets, T., Bakoev, F., Kolosov, A., Romanets, E., Korobeinikova, A., Bakoeva, I., Akhmedli, V., & Getmantseva, L. (2025). Visualization of Runs of Homozygosity and Classification Using Convolutional Neural Networks. Biology, 14(4), 426. https://doi.org/10.3390/biology14040426