
Genetic Diversity and Structure of Korean Pacific Oyster (Crassostrea gigas) for Determining Selective Breeding Groups

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction and Sampling
2.2. Microsatellite Loci Genotyping Analysis
2.3. Genetic Diversity Analysis
2.4. Genetic Structure of Populations Analysis
3. Results
3.1. Genetic Diversityof Wild Populations
3.2. Bottleneck Test and Effective Population Size Analysis
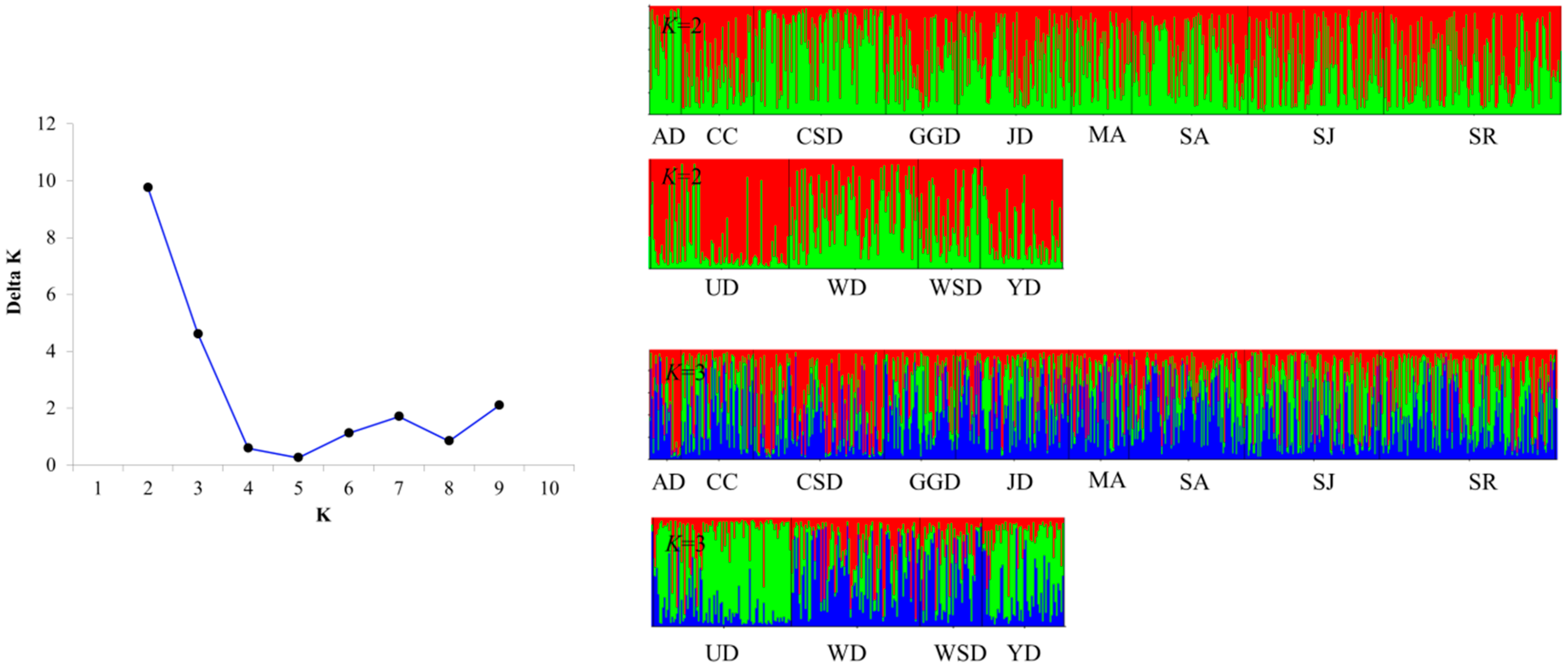
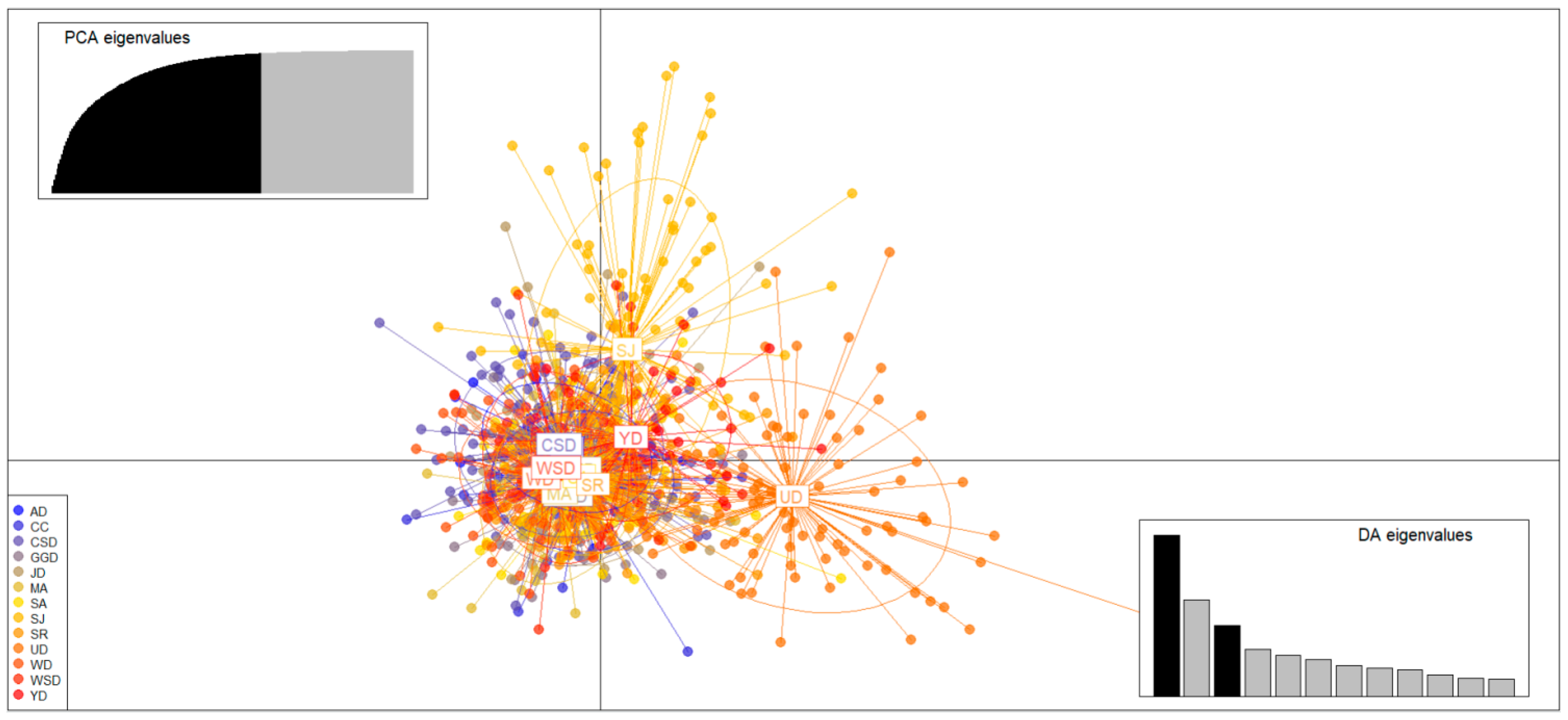
3.3. Genetic Structure and Genetic Differentiation of Wild Populations
4. Discussion
4.1. Genetic Diversity of Wild Populations
4.2. Genetic Structure of Wild Populations for C. gigas
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, D.Y.; Lee, G.Y. Direction of Restructuring in Response to Structural Changes in the Korean Oyster Farming. J. Korean Soc. Fish. Mar. Sci. Educ. 2022, 34, 404–414. [Google Scholar] [CrossRef]
- An, H.S.; Lee, J.W.; Kim, W.J.; Lim, H.J.; Kim, E.M.; Byun, S.G.; An, C.M. Comparative genetic diversity of wild and hatchery-produced Pacific oyster (Crassostrea gigas) populations in Korea using multiplex PCR assays with nine polymorphic microsatellite markers. Genes Genom. 2013, 35, 805–815. [Google Scholar] [CrossRef]
- An, H.S.; Kim, W.J.; Lim, H.J.; Byun, S.G.; Hur, Y.B.; Park, J.Y.; An, C.M. Genetic structure and diversity of Crassostrea gigas in Korea revealed from microsatellite markers. Biochem. Syst. Ecol. 2014, 55, 283–291. [Google Scholar] [CrossRef]
- Chi, Y.; Li, Q.; Liu, S.; Kong, L. Genetic parameters of growth and survival in the Pacific oyster Crassostrea gigas. Aquac. Res. 2021, 52, 282–290. [Google Scholar] [CrossRef]
- Kawamura, K.; Miyake, T.; Obata, M.; Aoki, H.; Komaru, A. Population Demography and Genetic Characteristics of the Pacific Oyster Crassostrea gigas in Japan. Biochem. Syst. Ecol. 2017, 70, 211–221. [Google Scholar] [CrossRef]
- Greenbaum, G.; Templeton, A.R.; Zarmi, Y.; Bar-David, S. Allelic richness following population founding events–a stochastic modeling framework incorporating gene flow and genetic drift. PLoS ONE 2014, 9, e115203. [Google Scholar] [CrossRef]
- Lallias, D.; Boudry, P.; Lapegue, S.; King, J.W.; Beaumont, A.R. Strategies for the Retention of High Genetic Variability in European Flat Oyster (Ostrea edulis) Restoration Programmes. Conserv. Genet. 2010, 11, 1899–1910. [Google Scholar] [CrossRef]
- Li, S.; Li, Q.; Yu, H.; Kong, L.; Liu, S. Genetic Variation and Population Structure of the Pacific Oyster Crassostrea gigas in the Northwestern Pacific Inferred from Mitochondrial COI Sequences. Fish. Sci. 2015, 81, 1071–1082. [Google Scholar] [CrossRef]
- An, H.S.; Park, K.J.; Cho, K.C.; Han, H.S.; Myeong, J.I. Genetic structure of Korean populations of the clam Ruditapes philippinarum inferred from microsatellite marker analysis. Biochem. Syst. Ecol. 2012, 44, 186–195. [Google Scholar] [CrossRef]
- Zheng, S.; Zhang, T.; Tu, K.; Li, L.; Liu, Z.; Wu, B.; Sun, X. Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology 2023, 12, 557. [Google Scholar] [CrossRef]
- Xu, L.; Li, Q.; Xu, C.; Yu, H.; Kong, L. Genetic Diversity and Effective Population Size in Successive Mass Selected Generations of Black Shell Strain Pacific Oyster (Crassostrea gigas) Based on Microsatellites and mtDNA Data. Aquaculture 2019, 500, 338–346. [Google Scholar] [CrossRef]
- Whitlock, R.; Stewart, G.B.; Goodman, S.J.; Piertney, S.B.; Butlin, R.K.; Pullin, A.S.; Burke, T. A Systematic Review of Phenotypic Responses to Between-Population Outbreeding. Environ. Evid. 2013, 2, 13. [Google Scholar] [CrossRef]
- Waser, N.M.; Price, M.V.; Shaw, R.G. Outbreeding Depression Varies among Cohorts of Ipomopsis aggregata Planted in Nature. Evolution 2000, 54, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Aulchenko, Y.S. Effects of Population Structure in Genome-Wide Association Studies. In Analysis of Complex Disease Association Studies; Academic Press: Cambridge, MA, USA, 2011; pp. 123–156. [Google Scholar]
- Wang, J.; Santiago, E.; Caballero, A. Prediction and estimation of effective population size. Heredity 2016, 117, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.M.; Lee, H.M.; Lee, M.N.; Noh, E.S.; Nam, B.H.; Kim, Y.O.; Kim, E.M. Development of new Microsatellite DNA Markers Using Next-generation Sequencing in Pacific Oyster Crassostrea gigas. Korean J. Malacol. 2022, 38, 129–138. [Google Scholar]
- Li, G.; Hubert, S.; Bucklin, K.; Ribes, V.; Hedgecock, D. Characterization of 79 microsatellite DNA markers in the Pacific oyster Crassostrea gigas. Mol. Ecol. Notes 2003, 3, 228–232. [Google Scholar] [CrossRef]
- Currie-Fraser, E.; Shah, P.; True, S. Data analysis using GeneMapper® v4.1: Comparing the newest generation of GeneMapper software to legacy Genescan® and Genotyper® Software. J. Biomol. Tech. 2010, 21, S31. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Marshall, T.C.; Slate, J.B.K.E.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. Population genetics software for exact test and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Piry, S.; Luikart, G.; Cornuet, J.M. Computer note. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Maruyama, T.; Fuerst, P.A. Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 1985, 111, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Li, Y.L.; Liu, J.X. StructureSelector: A web based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Kim, K.R.; Choi, H.K.; Lee, T.W.; Lee, H.J.; Yu, J.N. Population Structure and Genetic Diversity of the Spotted Sleeper Odontobutis interrupta (Odontobutidae), a Fish Endemic to Korea. Diversity 2023, 15, 913. [Google Scholar] [CrossRef]
- Kim, K.R.; Kwak, Y.H.; Sung, M.S.; Cho, S.J.; Bang, I.C. Population Structure and Genetic Diversity of the Endangered Fish Black Shinner Pseudopungtungia nigra (Cyprinidae) in Korea: A Wild and Restoration Population. Sci. Rep. 2023, 13, 9692. [Google Scholar] [CrossRef]
- Hong, Y.K.; Kim, K.R.; Kim, K.S.; Bang, I.C. The impact of Weir construction in Korea’s Nakdong River on the population genetic variability of the endangered fish species, Rapid small gudgeon (Microphysogobio rapidus). Genes 2023, 14, 1611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, C.; Li, Q. Genetic diversity in a genetically improved line of the Pacific oyster Crassostrea gigas with orange shell based on microsatellites and mtDNA data. Aquaculture 2022, 549, 737791. [Google Scholar] [CrossRef]
- Kim, K.R.; Park, S.Y.; Jeong, J.H.; Hwang, Y.; Kim, H.; Sung, M.S.; Yu, J.N. Genetic Diversity and Population Structure of Rhodeus uyekii in the Republic of Korea Revealed by Microsatellite Markers from Whole Genome Assembly. Int. J. Mol. Sci. 2024, 25, 6689. [Google Scholar] [CrossRef] [PubMed]
- De Meeûs, T. Revisiting FIS, FST, Wahlund effects, and null alleles. J. Hered. 2018, 109, 446–456. [Google Scholar] [CrossRef]
- Caballero, A.; Fernández, A.; Villanueva, B.; Toro, M.A. A comparison of marker-based estimators of inbreeding and inbreeding depression. Genet. Sel. Evol. 2022, 54, 82. [Google Scholar] [CrossRef]
- Bouzat, J.L. Conservation genetics of population bottlenecks: The role of chance, selection, and history. Conserv. Genet. 2010, 11, 463–478. [Google Scholar] [CrossRef]
- Geist, J.; Kuehn, R. Genetic diversity and differentiation of central European freshwater pearl mussel (Margaritifera margaritifera L.) populations: Implications for conservation and management. Mol. Ecol. 2005, 14, 425–439. [Google Scholar] [CrossRef]
- Alnajm, H.R.; Alijani, S.; Javanmard, A.; Rafat, A.; Hasanpur, K. Estimation of diversity structure, genetic bottleneck of three Iraqi sheep breeds using microsatellite markers. Anim. Environ. J. 2021, 13, 1–12. [Google Scholar] [CrossRef]
- Peery, M.Z.; Kirby, R.; Reid, B.N.; Stoelting, R.; Doucet-Bëer, E.L.; Robinson, S.; Palsbøll, P.J. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012, 21, 3403–3418. [Google Scholar] [CrossRef]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef]
- Doonan, J.; Beatty, G.E.; Sigwart, J.D.; Provan, J. Extensive local-scale gene flow and long-term population stability in the intertidal mollusc Katharina tunicata (Mollusca: Polyplacophora). Biol. J. Linn. Soc. 2012, 106, 589–597. [Google Scholar] [CrossRef]
- Bellgrove, A.; van Rooyen, A.; Weeks, A.R.; Clark, J.S.; Doblin, M.A.; Miller, A.D. New resource for population genetics studies on the Australasian intertidal brown alga, Hormosira banksii: Isolation and characterization of 15 polymorphic microsatellite loci through next generation DNA sequencing. J. Appl. Phycol. 2017, 29, 1721–1727. [Google Scholar] [CrossRef]
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.H.; Lawrence, E.R.; Matte, J.M.; Gallagher, B.K.; Salisbury, S.J.; Michaelides, S.N.; Fraser, D.J. Global assessment of effective population sizes: Consistent taxonomic differences in meeting the 50/500 rule. Mol. Ecol. 2024, 33, e17353. [Google Scholar] [CrossRef]
- Frankham, R.; Bradshaw, C.J.; Brook, B.W. Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]
- Stoeckle, B.C.; Araujo, R.; Geist, J.; Kuehn, R.; Toledo, C.; Machordom, A. Strong Genetic Differentiation and Low Genetic Diversity of the Freshwater Pearl Mussel (Margaritifera margaritifera L.) in the Southwestern European Distribution Range. Conserv. Genet. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Fraser, D.J. How well can captive breeding programs conserve biodiversity? A review of salmonids. Evol. Appl. 2008, 1, 535–586. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
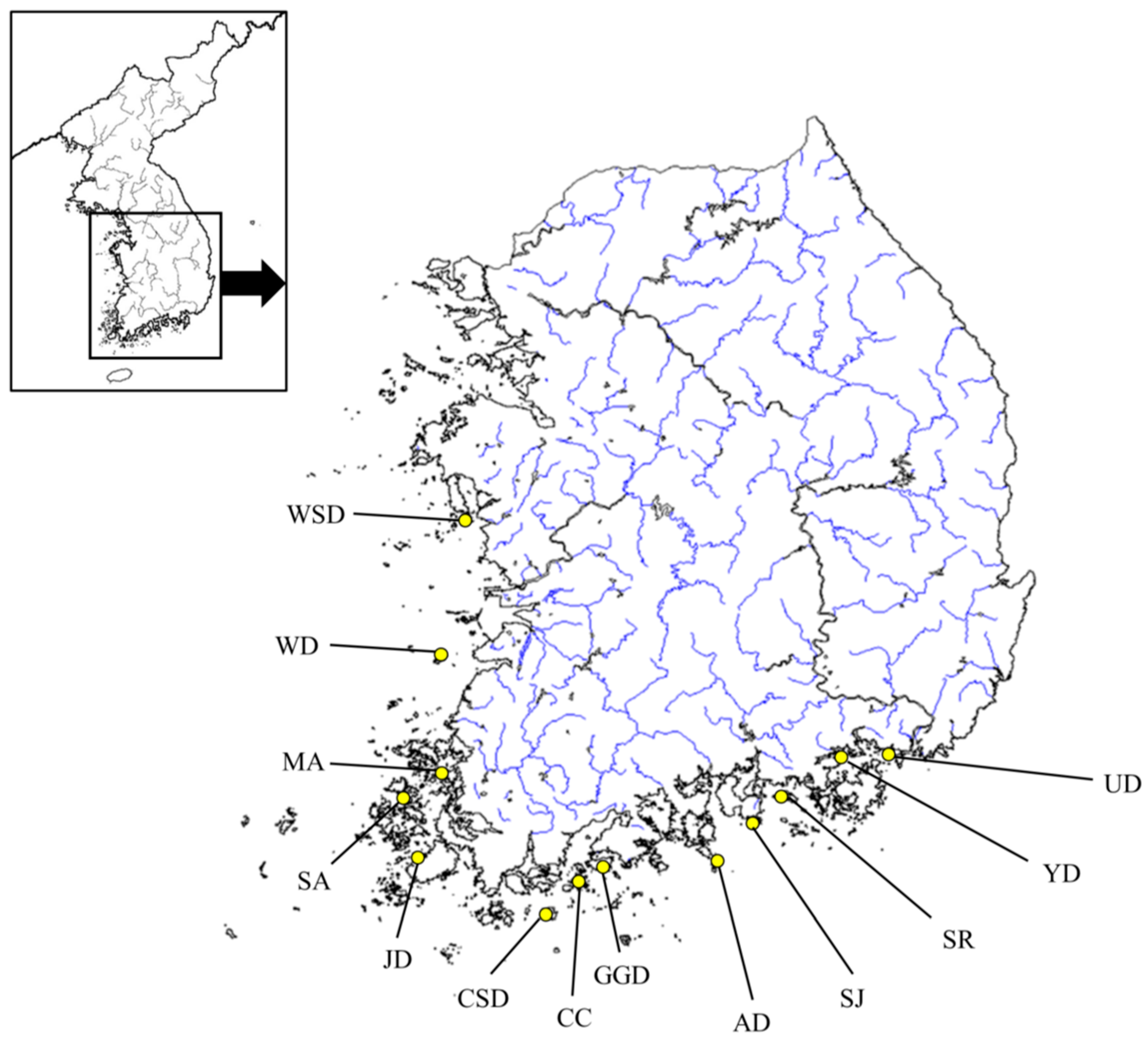
| Code | Location Name | Years | N | Location |
|---|---|---|---|---|
| AD | Ando | 2024 | 26 | 34°29′12″ N 127°48′17″ E |
| CC | Chukchi | 2024 | 60 | 34°22′14″ N 127°02′04″ E |
| CSD | Chungsando | 2024 | 109 | 34°10′48″ N 126°51′23″ E |
| GGD | Gugeumdo | 2024 | 59 | 34°29′13″ N 127°07′25″ E |
| JD | Jindo | 2024 | 94 | 34°25′18″ N 126°20′43″ E |
| MA | Muan | 2024 | 50 | 34°56′21″ N 126°23′25″ E |
| NDD | Nodaedo | 2024 | 25 | 34°40′09″ N 128°14′47″ E |
| SA | Sinan | 2024 | 96 | 34°51′58″ N 126°18′53″ E |
| SJ | Sangju | 2024 | 112 | 34°42′48″ N 127°59′12″ E |
| SR | Saryang | 2024 | 146 | 34°50′00″ N 128°08′13″ E |
| UD | Udo | 2024 | 89 | 35°05′16″ N 128°43′23″ E |
| WD | Wido | 2024 | 83 | 35°35′47″ N 126°16′04″ E |
| WSD | Wonsando | 2024 | 40 | 36°22′03″ N 126°23′42″ E |
| YD | Yangdo | 2024 | 53 | 35°04′55″ N 128°29′30″ E |
| ID | Location | N | NA | HO | HE | PHWE | FIS |
|---|---|---|---|---|---|---|---|
| AD | AnDo | 26 | 7.60 | 0.385 | 0.628 | 0.000 *** | 0.393 *** |
| CC | ChukChi | 60 | 9.60 | 0.404 | 0.593 | 0.000 *** | 0.318 *** |
| CSD | ChungSanDo | 109 | 11.07 | 0.406 | 0.600 | 0.000 *** | 0.319 *** |
| GGD | GuGeumDo | 59 | 9.13 | 0.393 | 0.585 | 0.000 *** | 0.325 *** |
| JD | JinDo | 94 | 10.60 | 0.339 | 0.580 | 0.000 *** | 0.417 *** |
| MA | MuAn | 50 | 9.00 | 0.397 | 0.605 | 0.000 *** | 0.345 *** |
| SA | SinAn | 96 | 10.47 | 0.418 | 0.593 | 0.000 *** | 0.297 *** |
| SJ | SangJu | 112 | 12.27 | 0.398 | 0.661 | 0.000 *** | 0.399 *** |
| SR | SaRyang | 146 | 12.07 | 0.412 | 0.596 | 0.000 *** | 0.310 *** |
| UD | UDo | 89 | 10.13 | 0.367 | 0.614 | 0.000 *** | 0.403 *** |
| WD | WiDo | 83 | 10.40 | 0.406 | 0.600 | 0.000 *** | 0.324 *** |
| WSD | WonSanDo | 40 | 8.60 | 0.395 | 0.592 | 0.000 *** | 0.335 *** |
| YD | YangDo | 53 | 9.00 | 0.413 | 0.591 | 0.000 *** | 0.301 *** |
| Population ID | N | Wilcoxon Sign-Rank Test | M Ratio | Ne | (95% CI) | |||
|---|---|---|---|---|---|---|---|---|
| PIAM | PTPM | PSMM | Mode-Shift | |||||
| AD | 26 | 0.000 *** | 0.268 | 0.542 | L-shaped | 0.333 | 28 | (21–37) |
| CC | 60 | 0.003 ** | 0.890 | 0.389 | L-shaped | 0.315 | 273 | (155–918) |
| CSD | 109 | 0.001 ** | 0.762 | 0.890 | L-shaped | 0.319 | 90 | (76–110) |
| GGD | 59 | 0.001 ** | 0.934 | 0.421 | L-shaped | 0.335 | 897 | (263–∞) |
| JD | 94 | 0.001 ** | 0.890 | 0.489 | L-shaped | 0.349 | 2491 | (539–∞) |
| MA | 50 | 0.000 *** | 0.389 | 0.978 | L-shaped | 0.333 | 388 | (183–∞) |
| SA | 96 | 0.000 *** | 0.542 | 0.542 | L-shaped | 0.367 | 1709 | (501–∞) |
| SJ | 112 | 0.002 ** | 0.978 | 0.303 | L-shaped | 0.374 | 441 | (295–823) |
| SR | 146 | 0.002 ** | 0.330 | 0.035 | L-shaped | 0.326 | 1381 | (607–∞) |
| UD | 89 | 0.002 ** | 0.561 | 0.890 | L-shaped | 0.358 | 314 | (206–610) |
| WD | 83 | 0.010 * | 0.890 | 0.229 | L-shaped | 0.330 | - | (2364–∞) |
| WSD | 40 | 0.000 *** | 0.358 | 0.761 | L-shaped | 0.357 | 1489 | (434–∞) |
| YD | 53 | 0.000 *** | 0.229 | 0.389 | L-shaped | 0.335 | - | (1210–∞) |
| AD | CC | CSD | GGD | JD | MA | SA | SJ | SR | UD | WD | WSD | YD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD | - | 0.002 | 0.152 | 0.014 | 0.000 | 0.002 | 0.000 | 0.000 | 0.000 | 0.000 | 0.008 | 0.006 | 0.000 |
| CC | 0.021 * | - | 0.000 | 0.618 | 0.646 | 0.392 | 0.797 | 0.000 | 0.654 | 0.000 | 0.406 | 0.557 | 0.015 |
| CSD | 0.008 | 0.018 * | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| GGD | 0.016 * | 0.002 | 0.015 * | - | 0.002 | 0.720 | 0.290 | 0.000 | 0.496 | 0.000 | 0.504 | 0.249 | 0.000 |
| JD | 0.023 * | 0.002 | 0.017 * | 0.009 * | - | 0.004 | 0.536 | 0.000 | 0.009 | 0.000 | 0.014 | 0.266 | 0.007 |
| MA | 0.020 ** | 0.004 | 0.016 * | 0.002 | 0.009 * | - | 0.285 | 0.000 | 0.020 | 0.000 | 0.055 | 0.293 | 0.000 |
| SA | 0.020 * | 0.001 | 0.013 * | 0.003 | 0.002 | 0.003 | - | 0.000 | 0.772 | 0.000 | 0.254 | 0.819 | 0.000 |
| SJ | 0.020 * | 0.013 * | 0.018 * | 0.015 * | 0.012 * | 0.015 * | 0.012 * | - | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| SR | 0.018 * | 0.001 | 0.013 * | 0.002 * | 0.004 * | 0.006 * | 0.000 | 0.012 * | - | 0.000 | 0.080 | 0.104 | 0.001 |
| UD | 0.057 * | 0.027 * | 0.047 * | 0.033 * | 0.036 * | 0.029 * | 0.029 * | 0.029 * | 0.029 * | - | 0.000 | 0.000 | 0.000 |
| WD | 0.015 * | 0.003 | 0.015 * | 0.002 * | 0.006 * | 0.006 | 0.002 | 0.013 * | 0.003 | 0.041 * | - | 0.381 | 0.000 |
| WSD | 0.021 * | 0.003 | 0.017 * | 0.005 | 0.005 | 0.005 | 0.001 | 0.014 * | 0.005 | 0.035 * | 0.003 | - | 0.000 |
| YD | 0.034 * | 0.009 * | 0.026 * | 0.019 * | 0.009 * | 0.021 * | 0.013 * | 0.014 * | 0.009 * | 0.026 * | 0.020 * | 0.020 * | - |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variance | F-Statistics |
|---|---|---|---|---|---|
| Microsatellite loci (One group based on the DAPC: AD, CC, CSD, GGD, JD, MA, SA, SJ, SR, UD, WD, WSD, and YD) | |||||
| Among groups | 12 | 176.028 | 0.05567 | 1.22 | 0.012 *** |
| Among populations within groups | 1004 | 6108.802 | 1.56091 | 34.09 | 0.345 *** |
| Within populations | 1017 | 3013.000 | 2.96264 | 64.70 | 0.353 *** |
| Total | 2033 | 9297.830 | 4.57922 | 100.00 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-R.; Lee, D.; Kim, K.-H.; Kim, H.C.; Kim, S.H.; Park, S.J.; Lee, D.-C. Genetic Diversity and Structure of Korean Pacific Oyster (Crassostrea gigas) for Determining Selective Breeding Groups. Biology 2025, 14, 449. https://doi.org/10.3390/biology14040449
Kim K-R, Lee D, Kim K-H, Kim HC, Kim SH, Park SJ, Lee D-C. Genetic Diversity and Structure of Korean Pacific Oyster (Crassostrea gigas) for Determining Selective Breeding Groups. Biology. 2025; 14(4):449. https://doi.org/10.3390/biology14040449
Chicago/Turabian StyleKim, Kang-Rae, Dain Lee, Kyung-Hee Kim, Hyun Chul Kim, So Hee Kim, Su Jin Park, and Deok-Chan Lee. 2025. "Genetic Diversity and Structure of Korean Pacific Oyster (Crassostrea gigas) for Determining Selective Breeding Groups" Biology 14, no. 4: 449. https://doi.org/10.3390/biology14040449
APA StyleKim, K.-R., Lee, D., Kim, K.-H., Kim, H. C., Kim, S. H., Park, S. J., & Lee, D.-C. (2025). Genetic Diversity and Structure of Korean Pacific Oyster (Crassostrea gigas) for Determining Selective Breeding Groups. Biology, 14(4), 449. https://doi.org/10.3390/biology14040449