
Animal Models in Rheumatoid Arthritis: Is There a Correlation Between Autoantibodies in Human Pathology and Animal Models?

 , , , , and
, , , , and
Simple Summary
Abstract

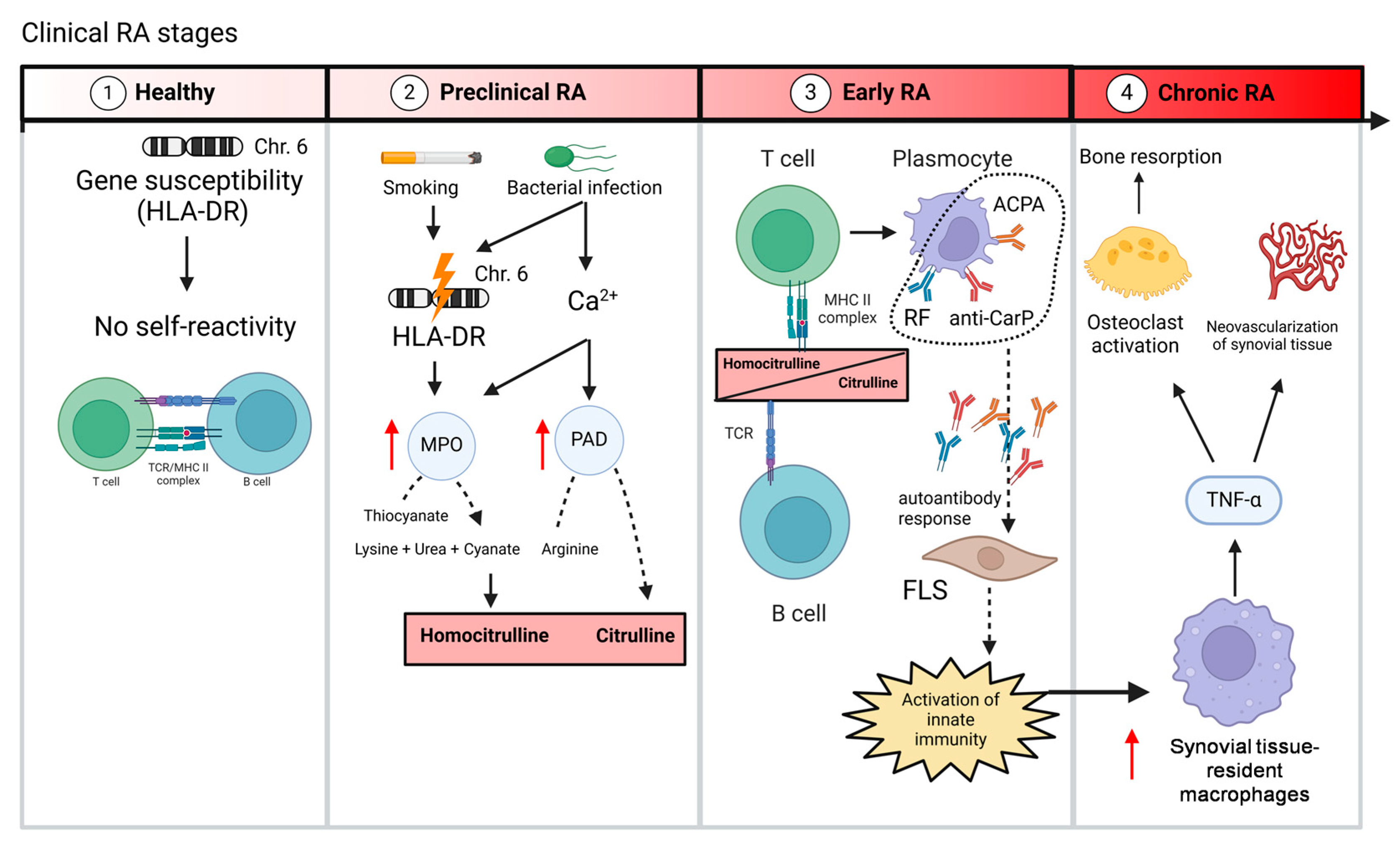
1. Introduction
2. Rheumatoid Factor in RA Patients
3. ACPA in RA Patients
4. Anti-Car-P in RA Patients
5. Other Autoantibodies Detected in RA Patients
6. Autoantibodies in Animal Models of RA
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAPAs | anti-acetylated protein antibodies |
| ACPA | anti-citrullinated protein antibodies |
| AIA | adjuvant-induced arthritis |
| ANA | anti-nuclear antibody |
| anti-CarP | anti-carbamylated protein antibodies |
| CAIA | collagen antibody-induced arthritis |
| CIA | collagen-induced arthritis |
| G6PI | glucose-6-phosphate isomerase |
| HLA | human leukocyte antigen |
| hTNFtg | TNF transgenic mice |
| NET | neutrophil extracellular trap |
| PAD | protein arginine deiminase enzymes |
| PIA | pristine-induced arthritis |
| RA | Rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| RF | rheumatoid factor |
References
- Gravallese Ellen, M.; Firestein Gary, S.; Natalie, K.; Emily, L.; Longo Dan, L.; Messenger Lori, A. Schubach Abigail What Is Rheumatoid Arthritis? N. Engl. J. Med. 2024, 390, e32. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Mechanisms of Joint Destruction in Rheumatoid Arthritis—Immune Cell–Fibroblast–Bone Interactions. Nat. Rev. Rheumatol. 2022, 18, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Sumida, T.S.; Cheru, N.T.; Hafler, D.A. The Regulation and Differentiation of Regulatory T Cells and Their Dysfunction in Autoimmune Diseases. Nat. Rev. Immunol. 2024, 24, 503–517. [Google Scholar] [CrossRef]
- Yang, M.; Zhu, L. Osteoimmunology: The Crosstalk between T Cells, B Cells, and Osteoclasts in Rheumatoid Arthritis. Int. J. Mol. Sci. 2024, 25, 2688. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef]
- Edilova, M.I.; Akram, A.; Abdul-Sater, A.A. Innate Immunity Drives Pathogenesis of Rheumatoid Arthritis. Biomed. J. 2021, 44, 172–182. [Google Scholar] [CrossRef]
- Prieto-Potin, I.; Largo, R.; Roman-Blas, J.A.; Herrero-Beaumont, G.; Walsh, D.A. Characterization of Multinucleated Giant Cells in Synovium and Subchondral Bone in Knee Osteoarthritis and Rheumatoid Arthritis. BMC Musculoskelet Disord 2015, 16, 226. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef]
- Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice. Cells 2020, 9, 1127. [Google Scholar] [CrossRef]
- Chang, M.H.; Nigrovic, P.A. Antibody-Dependent and -Independent Mechanisms of Inflammatory Arthritis. JCI Insight 2019, 4, e125278. [Google Scholar] [CrossRef]
- Kong, J.-S.; Jeong, G.H.; Yoo, S.-A. The Use of Animal Models in Rheumatoid Arthritis Research. J. Yeungnam Med. Sci. 2023, 40, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Xie, Z.; Xi, Y.; Liu, L.; Li, Z.; Qin, D. How to Model Rheumatoid Arthritis in Animals: From Rodents to Non-Human Primates. Front. Immunol. 2022, 13, 887460. [Google Scholar] [CrossRef]
- Iyengar, K.P.; Vaish, A.; Nune, A. Anti-Cyclic Citrullinated Peptide Antibody (ACPA) and Rheumatoid Arthritis: Clinical Relevance. J. Clin. Orthop. Trauma 2022, 24, 101729. [Google Scholar] [CrossRef] [PubMed]
- Fournier, C. Where Do T Cells Stand in Rheumatoid Arthritis? Jt. Bone Spine 2005, 72, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Stoop, J.N.; Liu, B.-S.; Shi, J.; Jansen, D.T.S.L.; Hegen, M.; Huizinga, T.W.J.; Trouw, L.A.; Toes, R.E.M. Antibodies Specific for Carbamylated Proteins Precede the Onset of Clinical Symptoms in Mice with Collagen Induced Arthritis. PLoS ONE 2014, 9, e102163. [Google Scholar] [CrossRef]
- Stoop, J.N.; Fischer, A.; Hayer, S.; Hegen, M.; Huizinga, T.W.; Steiner, G.; Trouw, L.A.; Toes, R.E. Anticarbamylated Protein Antibodies Can Be Detected in Animal Models of Arthritis That Require Active Involvement of the Adaptive Immune System. Ann. Rheum. Dis. 2015, 74, 949–950. [Google Scholar] [CrossRef]
- Gioud-Paquet, M.; Auvinet, M.; Raffin, T.; Girard, P.; Bouvier, M.; Lejeune, E.; Monier, J.C. IgM Rheumatoid Factor (RF), IgA RF, IgE RF, and IgG RF Detected by ELISA in Rheumatoid Arthritis. Ann. Rheum. Dis. 1987, 46, 65–71. [Google Scholar] [CrossRef]
- Maibom-Thomsen, S.L.; Trier, N.H.; Holm, B.E.; Hansen, K.B.; Rasmussen, M.I.; Chailyan, A.; Marcatili, P.; Højrup, P.; Houen, G. Immunoglobulin G Structure and Rheumatoid Factor Epitopes. PLoS ONE 2019, 14, e0217624. [Google Scholar] [CrossRef]
- Pike, R.M.; Sulkin, S.E.; Coggeshall, H.C. Serological Reactions in Rheumatoid Arthritis: I. Factors Affecting the Agglutination of Sensitized Sheep Erythrocytes in Rheumatoid-Arthritis Serum1. J. Immunol. 1949, 63, 441–446. [Google Scholar] [CrossRef]
- Wöhler, F.; Müller, W.; Hofmann, A. On the Nature of the Rheumatoid Factor. Ann. Rheum Dis. 1960, 19, 163. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; Mcshane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The American Rheumatism Association 1987 Revised Criteria for the Classification of Rheumatoid Arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef]
- Cerny, E.H.; Farshy, C.E.; Hunter, E.F.; Larsen, S.A. Rheumatoid Factor in Syphilis. J. Clin. Microbiol 1985, 22, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Elagib, K.E.E.; Børretzen, M.; Jonsson, R.; Haga, H.J.; Thoen, J.; Thompson, K.M.; Natvig, J.B. Rheumatoid Factors in Primary Sjögren’s Syndrome (pSS) Use Diverse VH Region Genes, the Majority of Which Show No Evidence of Somatic Hypermutation. Clin. Exp. Immunol. 2001, 117, 388–394. [Google Scholar] [CrossRef]
- Elkayam, O.; Segal, R.; Lidgi, M.; Caspi, D. Positive Anti-Cyclic Citrullinated Proteins and Rheumatoid Factor during Active Lung Tuberculosis. Ann. Rheum. Dis. 2006, 65, 1110–1112. [Google Scholar] [CrossRef]
- Radovanović-Dinić, B.; Tešić-Rajković, S.; Zivkovic, V.; Grgov, S. Clinical Connection between Rheumatoid Arthritis and Liver Damage. Rheumatol Int. 2018, 38, 715–724. [Google Scholar] [CrossRef]
- Serhal, L.; Lwin, M.N.; Holroyd, C.; Edwards, C.J. Rheumatoid Arthritis in the Elderly: Characteristics and Treatment Considerations. Autoimmun. Rev. 2020, 19, 102528. [Google Scholar] [CrossRef]
- Witte, T.; Hartung, K.; Sachse, C.; Matthias, T.; Fricke, M.; Kalden, J.R.; Lakomek, H.J.; Peter, H.H.; Schmidt, R.E. Rheumatoid Factors in Systemic Lupus Erythematosus: Association with Clinical and Laboratory Parameters. Rheumatol. Int. 2000, 19, 107–111. [Google Scholar] [CrossRef]
- Nicolò, A.; Amendt, T.; El Ayoubi, O.; Young, M.; Finzel, S.; Senel, M.; Voll, R.E.; Jumaa, H. Rheumatoid Factor IgM Autoantibodies Control IgG Homeostasis. Front. Immunol. 2022, 13, 1016263. [Google Scholar] [CrossRef]
- Motta, F.; Bizzaro, N.; Giavarina, D.; Franceschini, F.; Infantino, M.; Palterer, B.; Sebastiani, G.D.; Selmi, C. Rheumatoid Factor Isotypes in Rheumatoid Arthritis Diagnosis and Prognosis: A Systematic Review and Meta-Analysis. RMD Open 2023, 9, e002817. [Google Scholar] [CrossRef]
- Pertsinidou, E.; Saevarsdottir, S.; Manivel, V.A.; Klareskog, L.; Alfredsson, L.; Mathsson-Alm, L.; Hansson, M.; Cornillet, M.; Serre, G.; Holmdahl, R.; et al. In Early Rheumatoid Arthritis, Anticitrullinated Peptide Antibodies Associate with Low Number of Affected Joints and Rheumatoid Factor Associates with Systemic Inflammation. Ann. Rheum. Dis. 2024, 83, 277–287. [Google Scholar] [CrossRef]
- Sokolove, J.; Johnson, D.S.; Lahey, L.J.; Wagner, C.A.; Cheng, D.; Thiele, G.M.; Michaud, K.; Sayles, H.; Reimold, A.M.; Caplan, L.; et al. Rheumatoid Factor as a Potentiator of Anti–Citrullinated Protein Antibody–Mediated Inflammation in Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 813–821. [Google Scholar] [CrossRef]
- Aggarwal, R.; Liao, K.; Nair, R.; Ringold, S.; Costenbander, K.H. Anti–Citrullinated Peptide Antibody Assays and Their Role in the Diagnosis of Rheumatoid Arthritis. Arthritis Rheum. 2009, 61, 1472–1483. [Google Scholar] [CrossRef]
- Shi, J.; Van Steenbergen, H.W.; Van Nies, J.A.B.; Levarht, E.W.N.; Huizinga, T.W.J.; Van Der Helm-van Mil, A.H.M.; Toes, R.E.M.; Trouw, L.A. The Specificity of Anti-Carbamylated Protein Antibodies for Rheumatoid Arthritis in a Setting of Early Arthritis. Arthritis Res. Ther. 2015, 17, 339. [Google Scholar] [CrossRef] [PubMed]
- Studenic, P.; Alunno, A.; Sieghart, D.; Bang, H.; Aletaha, D.; Blüml, S.; Haslacher, H.; Smolen, J.S.; Gerli, R.; Steiner, G. Presence of Anti-Acetylated Peptide Antibodies (AAPA) in Inflammatory Arthritis and Other Rheumatic Diseases Suggests Discriminative Diagnostic Capacity towards Early Rheumatoid Arthritis. Ther. Adv. Musculoskelet. 2021, 13, 1759720X211022533. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Ge, H.; Dong, J.; Zhu, X.; Sun, G.; Ouyang, W.; Wang, L.; Zhang, G. The Diagnostic Significance of Glucose-6-Phosphate Isomerase (G6PI) Antigen and Anti-G6PI Antibody in Rheumatoid Arthritis Patients. Adv. Biosci. Biotechnol. 2013, 04, 818–822. [Google Scholar] [CrossRef]
- Yoshida, M.; Tsuji, M.; Kurosaka, D.; Kurosaka, D.; Yasuda, J.; Ito, Y.; Nishizawa, T.; Yamada, A. Autoimmunity to Citrullinated Type II Collagen in Rheumatoid Arthritis. Mod. Rheumatol. 2006, 16, 276–281. [Google Scholar] [CrossRef]
- Kay, J.; Upchurch, K.S. ACR/EULAR 2010 Rheumatoid Arthritis Classification Criteria. Rheumatology 2012, 51, vi5–vi9. [Google Scholar] [CrossRef]
- He, Y.; Ge, C.; Moreno-Giró, À.; Xu, B.; Beusch, C.M.; Sandor, K.; Su, J.; Cheng, L.; Lönnblom, E.; Lundqvist, C.; et al. A Subset of Antibodies Targeting Citrullinated Proteins Confers Protection from Rheumatoid Arthritis. Nat. Commun 2023, 14, 691. [Google Scholar] [CrossRef]
- Curran, A.M.; Naik, P.; Giles, J.T.; Darrah, E. PAD Enzymes in Rheumatoid Arthritis: Pathogenic Effectors and Autoimmune Targets. Nat. Rev. Rheumatol. 2020, 16, 301–315. [Google Scholar] [CrossRef]
- van Venrooij, W.J.; van Beers, J.J.B.C.; Pruijn, G.J.M. Anti-CCP Antibodies: The Past, the Present and the Future. Nat. Rev. Rheumatol. 2011, 7, 391–398. [Google Scholar] [CrossRef]
- Tilvawala, R.; Nguyen, S.H.; Maurais, A.J.; Nemmara, V.V.; Nagar, M.; Salinger, A.J.; Nagpal, S.; Weerapana, E.; Thompson, P.R. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chem. Biol. 2018, 25, 691–704.e6. [Google Scholar] [CrossRef]
- Foulquier, C.; Sebbag, M.; Clavel, C.; Chapuy-Regaud, S.; Al Badine, R.; Méchin, M.-C.; Vincent, C.; Nachat, R.; Yamada, M.; Takahara, H.; et al. Peptidyl Arginine Deiminase Type 2 (PAD-2) and PAD-4 but Not PAD-1, PAD-3, and PAD-6 Are Expressed in Rheumatoid Arthritis Synovium in Close Association with Tissue Inflammation. Arthritis Rheum. 2007, 56, 3541–3553. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, P.; Guo, S.; Schrodi, S.J.; He, D. Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 809806. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.A.; Wigerblad, G.; Sahlström, P.; Garimella, M.G.; Chemin, K.; Steen, J.; Titcombe, P.J.; Marklein, B.; Zhou, D.; Stålesen, R.; et al. Differential ACPA Binding to Nuclear Antigens Reveals a PAD-Independent Pathway and a Distinct Subset of Acetylation Cross-Reactive Autoantibodies in Rheumatoid Arthritis. Front. Immunol. 2019, 9, 3033. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Pratesi, F.; Prediletto, E.; Bombardieri, M.; Migliorini, P. NETosis as Source of Autoantigens in Rheumatoid Arthritis. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Reis, L.R.; Souza Junior, D.R.; Tomasin, R.; Bruni-Cardoso, A.; Di Mascio, P.; Ronsein, G.E. Citrullination of Actin-Ligand and Nuclear Structural Proteins, Cytoskeleton Reorganization and Protein Redistribution across Cellular Fractions Are Early Events in Ionomycin-Induced NETosis. Redox Biol. 2023, 64, 102784. [Google Scholar] [CrossRef]
- Liu, J.; Gao, J.; Wu, Z.; Mi, L.; Li, N.; Wang, Y.; Peng, X.; Xu, K.; Wu, F.; Zhang, L. Anti-Citrullinated Protein Antibody Generation, Pathogenesis, Clinical Application, and Prospects. Front. Med. 2022, 8, 802934. [Google Scholar] [CrossRef]
- Cîrciumaru, A.; Kisten, Y.; Hansson, M.; Mathsson-Alm, L.; Joshua, V.; Wähämaa, H.; Loberg Haarhaus, M.; Lindqvist, J.; Padyukov, L.; Catrina, S.-B.; et al. Identification of Early Risk Factors for Anti-Citrullinated-Protein-Antibody Positive Rheumatoid Arthritis—A Prospective Cohort Study. Rheumatology 2024, 63, 3164–3171. [Google Scholar] [CrossRef]
- Kastbom, A.; Roos Ljungberg, K.; Ziegelasch, M.; Wetterö, J.; Skogh, T.; Martinsson, K. Changes in Anti-Citrullinated Protein Antibody Isotype Levels in Relation to Disease Activity and Response to Treatment in Early Rheumatoid Arthritis. Clin. Exp. Immunol. 2018, 194, 391–399. [Google Scholar] [CrossRef]
- Van Delft, M.A.M.; Verheul, M.K.; Burgers, L.E.; Derksen, V.F.A.M.; Van Der Helm-van Mil, A.H.M.; Van Der Woude, D.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. The Isotype and IgG Subclass Distribution of Anti-Carbamylated Protein Antibodies in Rheumatoid Arthritis Patients. Arthritis Res. Ther. 2017, 19, 190. [Google Scholar] [CrossRef]
- Laurent, L.; Clavel, C.; Anquetil, F.; Pasquali, J.-L.; Sebbag, M.; Serre, G. Cytokine Profile of Macrophages in Vitro Stimulated by ACPA Immune Complexes in the Presence or Absence of IgM Rheumatoid Factor. Ann. Rheum. Dis. 2011, 70, A38. [Google Scholar] [CrossRef]
- Kempers, A.C.; Nejadnik, M.R.; Rombouts, Y.; Ioan-Facsinay, A.; van Oosterhout, M.; Jiskoot, W.; Huizinga, T.W.J.; Toes, R.E.M.; Scherer, H.U. Fc Gamma Receptor Binding Profile of Anti-Citrullinated Protein Antibodies in Immune Complexes Suggests a Role for FcγRI in the Pathogenesis of Synovial Inflammation. Clin. Exp. Rheumatol. 2018, 36, 284–293. [Google Scholar]
- Li, J.; Liu, P.; Huang, Y.; Wang, Y.; Zhao, J.; Xiong, Z.; Liu, M.; Wu, R. Immunophenotypic Landscape of Synovial Tissue in Rheumatoid Arthritis: Insights from ACPA Status. Heliyon 2024, 10, e34088. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, W.; Slowinska, I.; Krogulec, Z.; Syrowka, P.; Maslinski, W. Antibodies to Citrullinated Proteins (ACPA) Associate with Markers of Osteoclast Activation and Bone Destruction in the Bone Marrow of Patients with Rheumatoid Arthritis. J. Clin. Med. 2021, 10, 1778. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.Y.; Hur, B.; Gupta, V.K.; Arment, C.A.; Wright, K.A.; Mason, T.G.; Peterson, L.S.; Bekele, D.I.; Schaffer, D.E.; Bailey, M.L.; et al. Patients with ACPA-Positive and ACPA-Negative Rheumatoid Arthritis Show Different Serological Autoantibody Repertoires and Autoantibody Associations with Disease Activity. Sci. Rep. 2023, 13, 5360. [Google Scholar] [CrossRef] [PubMed]
- Burgers, L.E.; Van Steenbergen, H.W.; Ten Brinck, R.M.; Huizinga, T.W.; Van Der Helm-van Mil, A.H. Differences in the Symptomatic Phase Preceding ACPA-Positive and ACPA-Negative RA: A Longitudinal Study in Arthralgia during Progression to Clinical Arthritis. Ann. Rheum. Dis. 2017, 76, 1751–1754. [Google Scholar] [CrossRef]
- Pratesi, F.; Petit Teixeira, E.; Sidney, J.; Michou, L.; Puxeddu, I.; Sette, A.; Cornelis, F.; Migliorini, P. HLA Shared Epitope and ACPA: Just a Marker or an Active Player? Autoimmun. Rev. 2013, 12, 1182–1187. [Google Scholar] [CrossRef]
- Roudier, J.; Balandraud, N.; Auger, I. How RA Associated HLA-DR Molecules Contribute to the Development of Antibodies to Citrullinated Proteins: The Hapten Carrier Model. Front. Immunol. 2022, 13, 930112. [Google Scholar] [CrossRef]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.M.; Schreuder, G.M.T.; Breedveld, F.C.; Huizinga, T.W.J.; de Vries, R.R.P.; Toes, R.E.M. Association of HLA–DR3 with Anti–Cyclic Citrullinated Peptide Antibody–Negative Rheumatoid Arthritis. Arthritis Rheum. 2005, 52, 3058–3062. [Google Scholar] [CrossRef]
- Kampstra, A.S.B.; Toes, R.E.M. HLA Class II and Rheumatoid Arthritis: The Bumpy Road of Revelation. Immunogenetics 2017, 69, 597–603. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; De Moel, E.; Smolik, I.; Tanner, S.; Meng, X.; Jansen, B.C.; Bondt, A.; Wuhrer, M.; Huizinga, T.W.J.; Toes, R.E.M.; et al. N -Linked Glycans in the Variable Domain of IgG Anti–Citrullinated Protein Antibodies Predict the Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1626–1633. [Google Scholar] [CrossRef]
- Hemon, M.F.; Lambert, N.C.; Roudier, J.; Auger, I. PAD2 Immunization Induces ACPA in Wild-type and HLA-DR4 Humanized Mice. Eur. J. Immunol 2022, 52, 1464–1473. [Google Scholar] [CrossRef]
- Van Der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.M.; Huizinga, T.W.J.; Thomson, W.; Worthington, J.; Van Der Helm-van Mil, A.H.M.; De Vries, R.R.P. Quantitative Heritability of Anti–Citrullinated Protein Antibody–Positive and Anti–Citrullinated Protein Antibody–Negative Rheumatoid Arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Feitsma, A.L.; Toes, R.E.M.; Begovich, A.B.; Chokkalingam, A.P.; De Vries, R.R.P.; Huizinga, T.W.J.; Van Der Helm-van Mil, A.H.M. Risk of Progression from Undifferentiated Arthritis to Rheumatoid Arthritis: The Effect of the PTPN22 1858T-Allele in Anti-Citrullinated Peptide Antibody Positive Patients. Rheumatology 2007, 46, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Massarenti, L.; Enevold, C.; Damgaard, D.; Ødum, N.; Garred, P.; Frisch, M.; Shelef, M.A.; Jacobsen, S.; Nielsen, C.H. PADI4 Polymorphisms Confer Risk of Anti-CCP-Positive Rheumatoid Arthritis in Synergy With HLA-DRB1*04 and Smoking. Front. Immunol. 2021, 12, 707690. [Google Scholar] [CrossRef]
- Sparks, J.A.; Karlson, E.W. The Roles of Cigarette Smoking and the Lung in the Transitions Between Phases of Preclinical Rheumatoid Arthritis. Curr. Rheumatol Rep. 2016, 18, 15. [Google Scholar] [CrossRef]
- Ahmadi, P.; Mahmoudi, M.; Kheder, R.K.; Faraj, T.A.; Mollazadeh, S.; Abdulabbas, H.S.; Esmaeili, S.-A. Impacts of Porphyromonas Gingivalis Periodontitis on Rheumatoid Arthritis Autoimmunity. Int. Immunopharmacol. 2023, 118, 109936. [Google Scholar] [CrossRef]
- Kwon, E.-J.; Ju, J.H. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. Int. J. Mol. Sci. 2021, 22, 10576. [Google Scholar] [CrossRef]
- Darrah, E.; Andrade, F. Rheumatoid Arthritis and Citrullination. Curr. Opin. Rheumatol. 2018, 30, 72–78. [Google Scholar] [CrossRef]
- Pruijn, G.J.M. Citrullination and Carbamylation in the Pathophysiology of Rheumatoid Arthritis. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Jiang, X.; Trouw, L.A.; van Wesemael, T.J.; Shi, J.; Bengtsson, C.; Källberg, H.; Malmström, V.; Israelsson, L.; Hreggvidsdottir, H.; Verduijn, W.; et al. Anti-CarP Antibodies in Two Large Cohorts of Patients with Rheumatoid Arthritis and Their Relationship to Genetic Risk Factors, Cigarette Smoking and Other Autoantibodies. Ann. Rheum. Dis. 2014, 73, 1761–1768. [Google Scholar] [CrossRef]
- Dibrov, D.A.; Avdeeva, A.S.; Diatroptov, M.E.; Nasonov, E.L. Anti-Carbamylated Protein Antibodies in ACPA-Negative and ACPA-Positive Patients with Rheumatoid Arthritis. Dokl. Biochem. Biophys. 2024, 517, 235–242. [Google Scholar] [CrossRef]
- O’Neil, L.J.; Barrera-Vargas, A.; Sandoval-Heglund, D.; Merayo-Chalico, J.; Aguirre-Aguilar, E.; Aponte, A.M.; Ruiz-Perdomo, Y.; Gucek, M.; El-Gabalawy, H.; Fox, D.A.; et al. Neutrophil-Mediated Carbamylation Promotes Articular Damage in Rheumatoid Arthritis. Sci. Adv. 2020, 6, eabd2688. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, V.; Recalchi, S.; Capozzi, A.; Riitano, G.; Mattei, V.; Longo, A.; Di Franco, M.; Alessandri, C.; Bombardieri, M.; Valesini, G.; et al. Autophagy Induces Protein Carbamylation in Fibroblast-like Synoviocytes from Patients with Rheumatoid Arthritis. Rheumatology 2018, 57, 2032–2041. [Google Scholar] [CrossRef]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Krämer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of Vimentin Is Inducible by Smoking and Represents an Independent Autoantigen in Rheumatoid Arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef]
- Regueiro, C.; Rodríguez-Rodríguez, L.; Triguero-Martinez, A.; Nuño, L.; Castaño-Nuñez, A.L.; Villalva, A.; Pérez-Pampín, E.; Lopez-Golan, Y.; Abasolo, L.; Ortiz, A.M.; et al. Specific Association of HLA—DRB 1*03 with Anti–Carbamylated Protein Antibodies in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; van Veelen, P.A.; Mahler, M.; Janssen, G.M.C.; Drijfhout, J.W.; Huizinga, T.W.J.; Toes, R.E.M.; Trouw, L.A. Carbamylation and Antibodies against Carbamylated Proteins in Autoimmunity and Other Pathologies. Autoimmun. Rev. 2014, 13, 225–230. [Google Scholar] [CrossRef]
- Wynckel, A.; Randoux, C.; Millart, H.; Desroches, C.; Gillery, P.; Canivet, E.; Chanard, J. Kinetics of Carbamylated Haemoglobin in Acute Renal Failure. Nephrol. Dial. Transplant. 2000, 15, 1183–1188. [Google Scholar] [CrossRef]
- Arito, M.; Nagai, K.; Ooka, S.; Sato, T.; Takakuwa, Y.; Kurokawa, M.S.; Sase, T.; Okamoto, K.; Suematsu, N.; Kato, T. Altered Acetylation of Proteins in Patients with Rheumatoid Arthritis Revealed by Acetyl-Proteomics. Clin. Exp. Rheumatol. 2025, 33, 877–886. [Google Scholar]
- Kampstra, A.S.B.; Dekkers, J.S.; Volkov, M.; Dorjée, A.L.; Hafkenscheid, L.; Kempers, A.C.; van Delft, M.; Kissel, T.; Reijm, S.; Janssen, G.M.C.; et al. Different Classes of Anti-Modified Protein Antibodies Are Induced on Exposure to Antigens Expressing Only One Type of Modification. Ann. Rheum Dis. 2019, 78, 908. [Google Scholar] [CrossRef]
- Schaller, M.; Burton, D.R.; Ditzel, H.J. Autoantibodies to GPI in Rheumatoid Arthritis: Linkage between an Animal Model and Human Disease. Nat. Immunol. 2001, 2, 746–753. [Google Scholar] [CrossRef]
- Schaller, M.; Stohl, W.; Benoit, V.; Tan, S.-M.; Johansen, L.; Ditzel, H.J. Patients with Inflammatory Arthritic Diseases Harbor Elevated Serum and Synovial Fluid Levels of Free and Immune-Complexed Glucose-6-Phosphate Isomerase (G6PI). Biochem. Biophys. Res. Commun. 2006, 349, 838–845. [Google Scholar] [CrossRef]
- Cook, A.D.; Rowley, M.J.; Mackay, I.R.; Gough, A.; Emery, P. Antibodies to Type II Collagen in Early Rheumatoid Arthritis. Correlation with Disease Progression. Arthritis Rheum. 1996, 39, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Mullazehi, M.; Mathsson, L.; Lampa, J.; Rönnelid, J. High Anti-Collagen Type-II Antibody Levels and Induction of Proinflammatory Cytokines by Anti-Collagen Antibody-Containing Immune Complexes in Vitro Characterise a Distinct Rheumatoid Arthritis Phenotype Associated with Acute Inflammation at the Time of Disease Onset. Ann. Rheum. Dis. 2007, 66, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Largo, R.; Sánchez-Pernaute, O.; Marcos, M.E.; Moreno-Rubio, J.; Aparicio, C.; Granado, R.; Ortega, L.; Egido, J.; Herrero-Beaumont, G. Chronic Arthritis Aggravates Vascular Lesions in Rabbits with Atherosclerosis: A Novel Model of Atherosclerosis Associated with Chronic Inflammation. Arthritis Rheum. 2008, 58, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Little, R.D.; Prieto-Potin, I.; Pérez-Baos, S.; Villalvilla, A.; Gratal, P.; Cicuttini, F.; Largo, R.; Herrero-Beaumont, G. Compensatory Anabolic Signaling in the Sarcopenia of Experimental Chronic Arthritis. Sci. Rep. 2017, 7, 6311. [Google Scholar] [CrossRef]
- Pérez-Baos, S.; Barrasa, J.I.; Gratal, P.; Larrañaga-Vera, A.; Prieto-Potin, I.; Herrero-Beaumont, G.; Largo, R. Tofacitinib Restores the Inhibition of Reverse Cholesterol Transport Induced by Inflammation: Understanding the Lipid Paradox Associated with Rheumatoid Arthritis. Br. J. Pharmacol. 2017, 174, 3018–3031. [Google Scholar] [CrossRef]
- Kuhn, K.A. Antibodies against Citrullinated Proteins Enhance Tissue Injury in Experimental Autoimmune Arthritis. J. Clin. Investig. 2006, 116, 961–973. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Nijenhuis, S.; Helsen, M.M.A.; Van Der Heijden, A.; Senshu, T.; Van Den Berg, W.B.; Van Venrooij, W.J.; Joosten, L.A.B. Citrullination of Synovial Proteins in Murine Models of Rheumatoid Arthritis. Arthritis Rheum. 2003, 48, 2489–2500. [Google Scholar] [CrossRef]
- Kidd, B.A.; Ho, P.P.; Sharpe, O.; Zhao, X.; Tomooka, B.H.; Kanter, J.L.; Steinman, L.; Robinson, W.H. Epitope Spreading to Citrullinated Antigens in Mouse Models of Autoimmune Arthritis and Demyelination. Arthritis Res. Ther. 2008, 10, R119. [Google Scholar] [CrossRef]
- Kim, J.-W.; Jung, H.; Baek, I.-P.; Nam, Y.; Kang, J.; Chung, M.K.; Park, J.-B.; Lee, J.; Kwok, S.-K.; Kim, W.-U.; et al. Differential Effects of Periodontal Microbiome on the Rheumatoid Factor Induction during Rheumatoid Arthritis Pathogenesis. Sci. Rep. 2022, 12, 19636. [Google Scholar] [CrossRef]
- Zhao, X.; Long, J.; Liang, F.; Liu, N.; Sun, Y.; Xi, Y. Different Protective Efficacies of a Novel Antigen-Specific DNA Vaccine Encoding Chicken Type II Collagen via Intramuscular, Subcutaneous, and Intravenous Vaccination against Experimental Rheumatoid Arthritis. Biomed. Pharmacother. 2021, 144, 112294. [Google Scholar] [CrossRef]
- Verheul, M.K.; Vierboom, M.P.M.; ’T Hart, B.A.; Toes, R.E.M.; Trouw, L.A. Anti-Carbamylated Protein Antibodies Precede Disease Onset in Monkeys with Collagen-Induced Arthritis. Arthritis Res. Ther. 2017, 19, 246. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-D.; He, C.-Y.; Wu, Y.-J.; Xu, L.; Shi, C.; Olatunji, O.J.; Zuo, J.; Ji, C.-L. AMPK/SIRT1 Deficiency Drives Adjuvant-Induced Arthritis in Rats by Promoting Glycolysis-Mediated Monocytes Inflammatory Polarization. J. Inflamm. Res. 2022, 15, 4663–4675. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.A.; Ahmed, O.M.; Fahim, H.I.; Ali, T.M.; Elesawy, B.H.; Ashour, M.B. Potency of Bone Marrow-Derived Mesenchymal Stem Cells and Indomethacin in Complete Freund’s Adjuvant-Induced Arthritic Rats: Roles of TNF-α, IL-10, iNOS, MMP-9, and TGF-Β1. Stem Cells Int. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Hemmers, S.; Arandjelovic, S.; Corr, M.; Mowen, K.A. PAD4 Is Not Essential for Disease in the K/BxN Murine Autoantibody-Mediated Model of Arthritis. Arthritis Res. Ther. 2012, 14, R104. [Google Scholar] [CrossRef]
- Mancardi, D.; Jönsson, F.; Iannascoli, B.; Khun, H.; Van Rooijen, N.; Huerre, M.; Daëron, M.; Bruhns, P. Cutting Edge: The Murine High-Affinity IgG Receptor Fc Gamma RIV Is Sufficient for Autoantibody-Induced Arthritis. J. Immunol. 2011, 186, 1899–1903. [Google Scholar] [CrossRef]
- Maccioni, M.; Zeder-Lutz, G.; Huang, H.; Ebel, C.; Gerber, P.; Hergueux, J.; Marchal, P.; Duchatelle, V.; Degott, C.; Van Regenmortel, M.; et al. Arthritogenic Monoclonal Antibodies from K/BxN Mice. J. Exp. Med. 2002, 195, 1071–1077. [Google Scholar] [CrossRef]
- Piao, Y.; Yun, S.Y.; Kim, H.S.; Park, B.K.; Ha, H.C.; Fu, Z.; Jang, J.M.; Back, M.J.; Shin, I.C.; Won, J.H.; et al. A Novel Therapeutic Effect of a New Variant of CTLA4-Ig with Four Antennas That Are Terminally Capped with Sialic Acid in the CTLA4 Region. Biomol Ther. 2022, 30, 529–539. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Main Autoantibodies Produced in RA | ||||||
|---|---|---|---|---|---|---|
| RF | ACPA | Anti-CarP | AAPA | Anti-G6PI | Anti-Type II Collagen | |
| Specificity | For IgM-RF isotype the specificity was about 89.5% to 90.7% [29]. For IgA-RF isotype the specificity was about 90.8% to 92.0% [29]. | Specificity was about 85 to 99% [32]. | Specificity was about 89% [33]. | Specificity was about 77% [34]. | Specificity was about 93% [35]. | Specificity was about 95% [36]. |
| Sensitivity | For IgM-RF isotype the sensibility was about 62.1% to 64.6% [29]. For IgA-RF isotype the sensibility was about 48% to 50% [29]. | Sensibility was about 60 to 80% [32]. CCP2 assays have higher sensitivity than CCP1 assays [32]. | Sensibility was about 44% [33]. | Sensibility was about 60% for patients with early RA and about 68% for long-term RA [34]. | Sensibility was about 75% [35]. | Sensibility was about 79% [36]. |
| Pathogenic relevance | RF interacts with IgG antibodies to create immune complexes. These complexes tend to accumulate within the synovial joints, where they activate the complement system and initiate a cycle of chronic inflammation [29]. This promotes the infiltration of immune cells such as neutrophils and macrophages into the joint space. In turn, these cells release pro-inflammatory cytokines which intensify inflammation and contribute to the degradation of joint tissue and the development of synovitis [29]. | ACPA recognize and bind to citrullinated proteins leading to the formation of immune complexes. These complexes activate the complement system and engage Fc receptors on antigen-presenting cells like macrophages and dendritic cells. This immune activation triggers the release of pro-inflammatory cytokines which increases chronic synovial inflammation and promote joint tissue destruction in rheumatoid arthritis. In addition to their inflammatory role, ACPA have been shown to directly stimulate bone resorption [32]. | Anti-CarP antibodies recognize and attach to carbamylated proteins, leading to the formation of immune complexes. These complexes can accumulate in joint tissues, where they trigger activation of the complement system. This activation attracts macrophages and neutrophils and stimulates the production of pro-inflammatory cytokines such as TNF-α and IL-6. The resulting inflammatory environment contributes to synovial membrane damage and joint destruction [33]. | Mechanisms leading to the generation of AAPAs need to be characterized [34]. | Anti-G6PI antibodies interact with soluble or extracellular G6PI that is released during cellular stress or injury. This binding leads to the formation of immune complexes, which activate the complement system and trigger Fc receptor engagement on immune cells. This activates neutrophils, macrophages, and increases proinflammatory cytokines, developing synovial inflammation and joint degradation [35]. | Type II collagen is a key structural protein found in articular cartilage and is essential for maintaining joint integrity. In RA, the immune system target type II collagen, resulting in the production of anti-CII antibodies. These antibodies then bind to type II collagen, initiating an immune reaction that contributes to the degradation of cartilage [36]. |
| Diagnostic value | RF is detected in approximately 70 to 80% of RA patients, especially in those with more established or severe disease [29]. RF is included in the 2010 ACR/EULAR classification criteria for RA [37]. Higher titers of RF increase the likelihood of RA, particularly in combination with clinical symptoms and other antibodies like anti-CCP [29]. | ACPA can be detected years before the appearance of clinical symptoms [32]. ACPA are present in approximately 60 to 70% of individuals with RA [32]. ACPA is included in the 2010 ACR/EULAR classification criteria for RA [37]. | The anti-CarP are less sensitive than other markers, but its high specificity and presence in seronegative RA enhance its diagnostic value when used in combination with clinical findings [33]. Correlated with more severe prognosis of RA [33]. | AAPA can also be detected in a portion of seronegative patients for ACPA and anti-CarP, suggesting they may offer additional diagnostic insight [34]. | Useful in diagnosing seronegative RA patients, where RF and ACPA are not detected [35]. Correlated with more severe prognosis of RA [35]. | The anti-type II collagen has low specificity. Is found in other diseases, such as osteoarthritis and other autoimmune conditions [36]. Also is low sensitivity, is not found in the majority of RA patients seropositive for ACPA or RF [36]. |
| RA Model | Animal | Auto-Antibodies | Studies |
|---|---|---|---|
| CIA | Mice | ACPA − |
|
| |||
| ACPA + |
| ||
| |||
| Anti-Car-P + |
| ||
| |||
| RF + |
| ||
| Anti-collagen type II + |
| ||
| |||
| Rat | ACPA − |
| |
| |||
| Anti-Car-P + |
| ||
| RF − |
| ||
| Rhesus monkey | ACPA − |
| |
| Anti-Car-P + |
| ||
| RF − |
| ||
| AIA | Rat | ACPA − |
|
| |||
| Anti-Car-P + |
| ||
| RF − |
| ||
| PIA | Rat | ACPA − |
|
| Anti-Car-P + |
| ||
| hTNFtg | Mice | ACPA − |
|
| Anti-Car-P + |
| ||
| CAIA | Mice | ACPA − |
|
| Anti-Car-P + |
| ||
| K/BxN | Mice | Anti-GPI + |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marco-Bonilla, M.; Fresnadillo, M.; de la Riva-Bueno, M.; Herrero-Beaumont, G.; Largo, R.; Mediero, A. Animal Models in Rheumatoid Arthritis: Is There a Correlation Between Autoantibodies in Human Pathology and Animal Models? Biology 2025, 14, 460. https://doi.org/10.3390/biology14050460
Marco-Bonilla M, Fresnadillo M, de la Riva-Bueno M, Herrero-Beaumont G, Largo R, Mediero A. Animal Models in Rheumatoid Arthritis: Is There a Correlation Between Autoantibodies in Human Pathology and Animal Models? Biology. 2025; 14(5):460. https://doi.org/10.3390/biology14050460
Chicago/Turabian StyleMarco-Bonilla, Miguel, Maria Fresnadillo, Macarena de la Riva-Bueno, Gabriel Herrero-Beaumont, Raquel Largo, and Aránzazu Mediero. 2025. "Animal Models in Rheumatoid Arthritis: Is There a Correlation Between Autoantibodies in Human Pathology and Animal Models?" Biology 14, no. 5: 460. https://doi.org/10.3390/biology14050460
APA StyleMarco-Bonilla, M., Fresnadillo, M., de la Riva-Bueno, M., Herrero-Beaumont, G., Largo, R., & Mediero, A. (2025). Animal Models in Rheumatoid Arthritis: Is There a Correlation Between Autoantibodies in Human Pathology and Animal Models? Biology, 14(5), 460. https://doi.org/10.3390/biology14050460