
Evolutionary Origin and Genetic Diversity of the Pannonian Ecotype of Apis mellifera carnica Colonies in Hungary Based on Mitochondrial DNA and Microsatellite Markers

 ,
,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Mitochondrial DNA
2.4. Microsatellite Genotyping
2.5. Statistical Methods Used for Data Analysis
2.5.1. Mitochondrial DNA Analysis
2.5.2. Microsatellite Marker Analysis
3. Results
3.1. Mitochondrial DNA Information of the Honey Bee Groups Studied
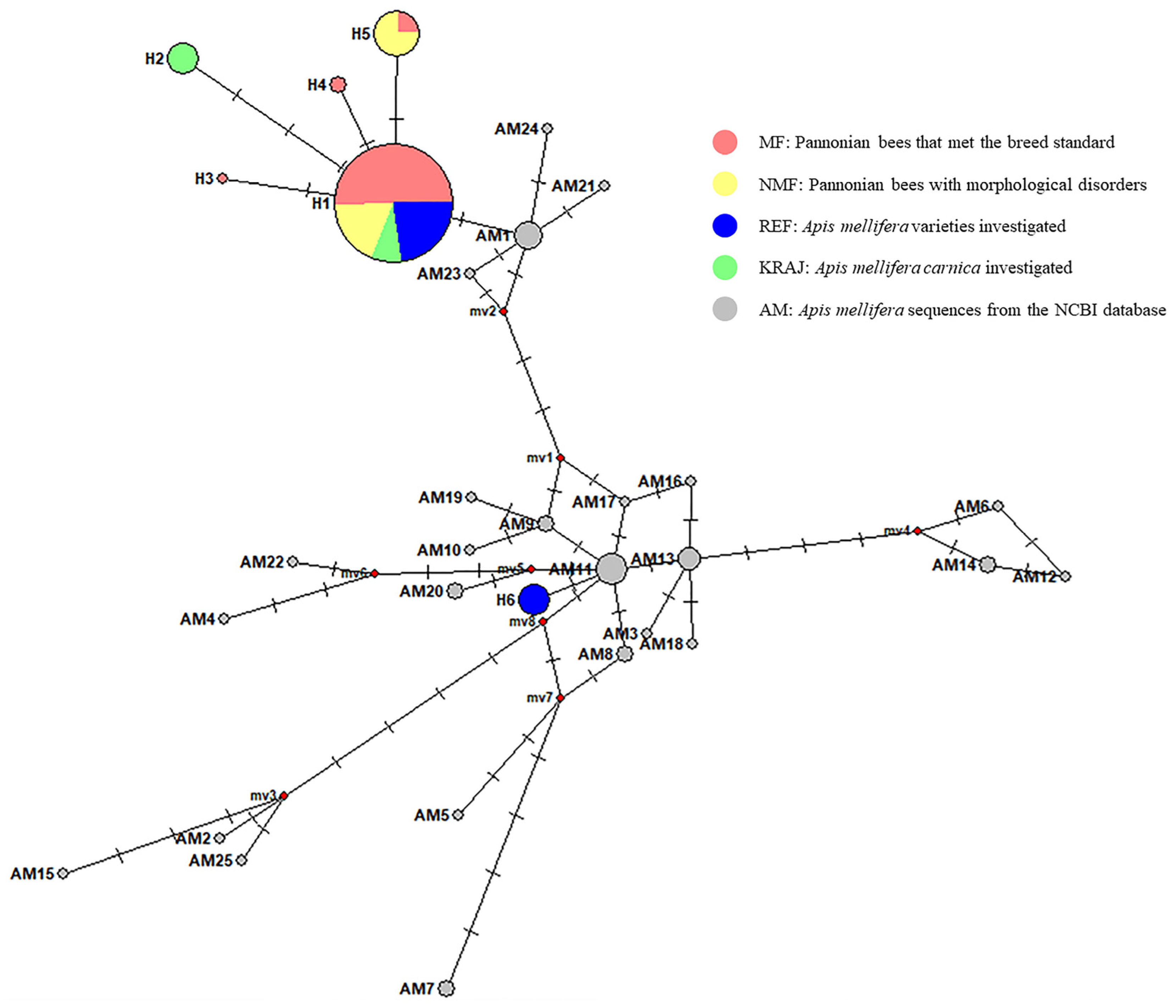
3.1.1. Genetic Variance of the Pannonian Bee Based on the COI and 16S mtDNA Regions
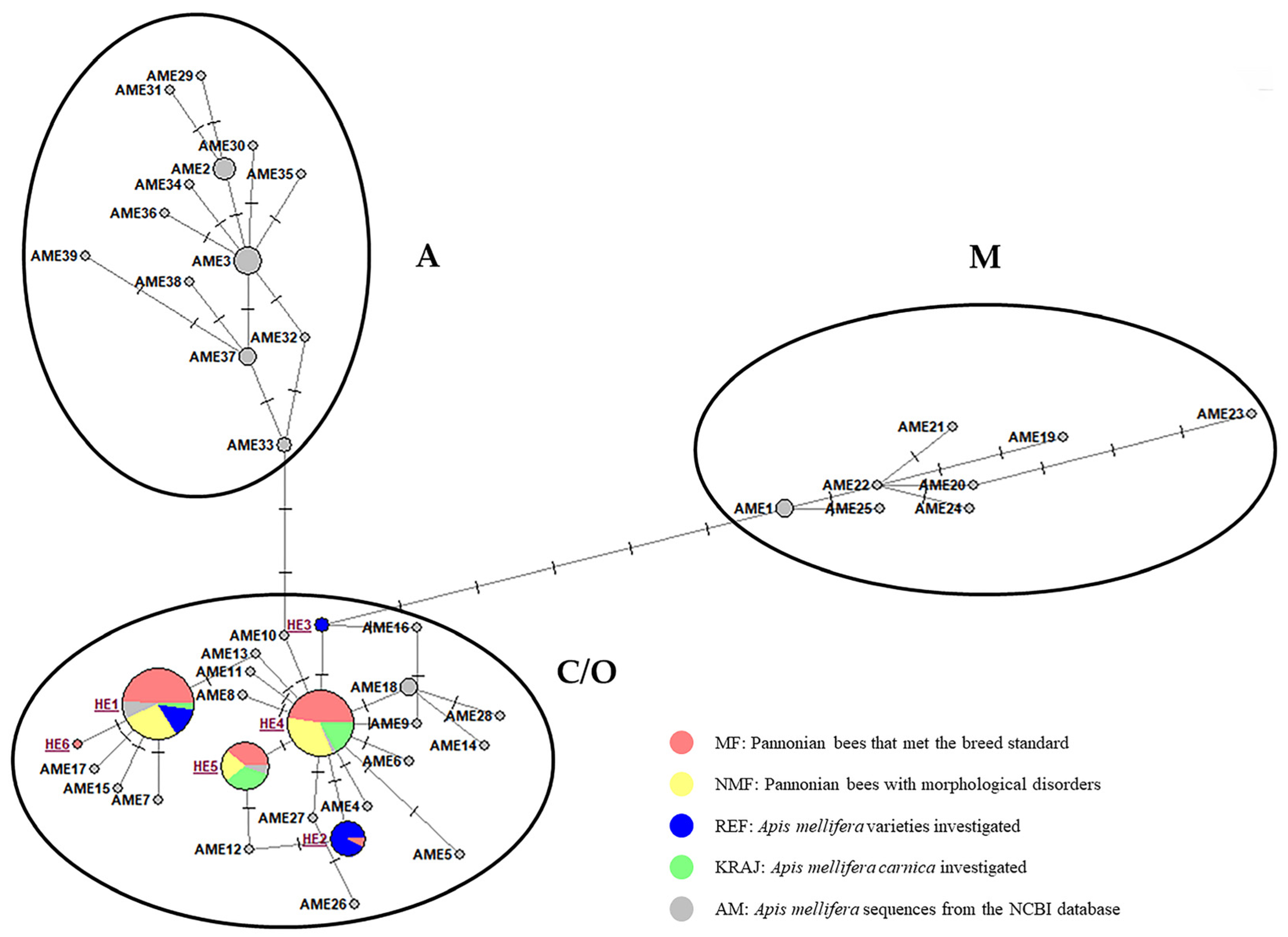
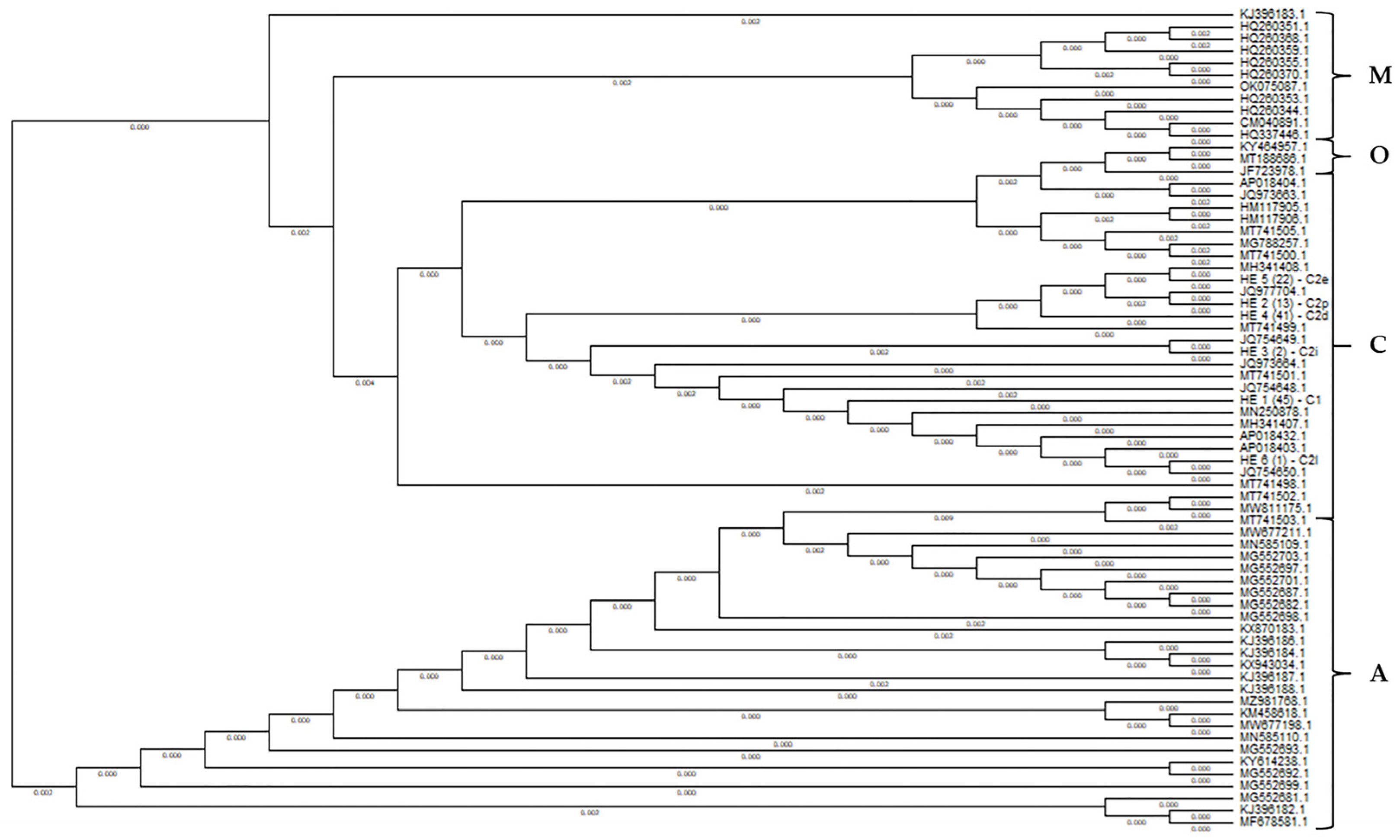
3.1.2. Evolutionary Origin of the Pannonian Bee in Hungary Based on the tRNAleu-Cox2 Intergenic Region (E2/H2)
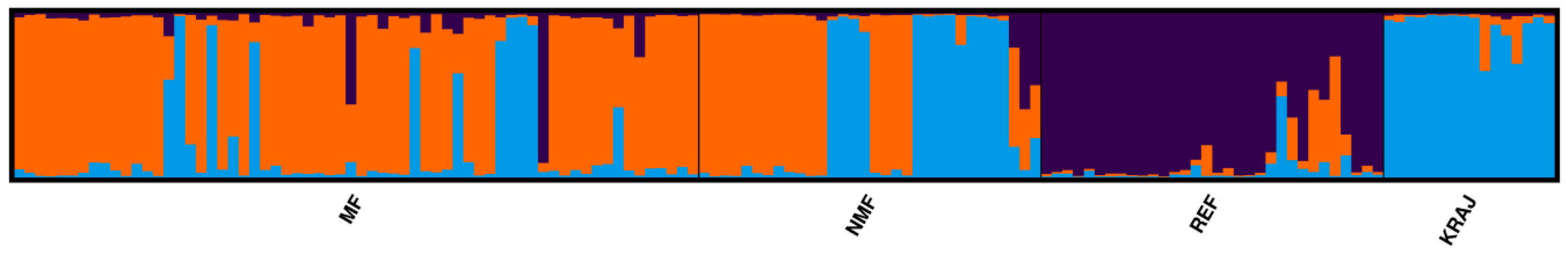
3.2. Microsatellite Marker Analysis of the Honey Bee Groups Investigated
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, A.M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of pollinators in changing landscapes for world crops. Proc. R. Soc. B 2007, 274, 303–313. [Google Scholar] [CrossRef]
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin/Heiderberg, Germany, 1988. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Lee, M.; Takahashi, J.; Wook Kwon, H.; Nikolenko, A.G. A revision of subspecies structure of western honey bee Apis mellifera. Saudi J. Biol. Sci. 2020, 27, 3615–3621. [Google Scholar] [CrossRef] [PubMed]
- vanEngelsdorp, D.; Hayes, J.; Underwood, R.M.; Pettis, J.S. A survey of honey bee colony losses in the United States, fall 2008 to spring 2009. J. Apic. Res. 2010, 49, 7–14. [Google Scholar] [CrossRef]
- Themudo, E.G.; Rey-Iglesia, A.; Tascón, R.L.; Jensen, A.B.; Fonseca, R.R.; Campos, P.F. Declining genetic diversity of European honeybees along the twentieth century. Sci. Rep. 2020, 10, 10520. [Google Scholar] [CrossRef]
- Potts, S.G.; Roberts, S.P.M.; Dean, R.; Marris, G.; Brown, M.A.; Jones, R.; Neumann, P.; Settele, J. Declines of managed honey bees and beekeepers in Europe. J. Apic. Res. 2010, 49, 15–22. [Google Scholar] [CrossRef]
- Stokstad, E. ENTOMOLOGY: The Case of the Empty Hives. Science 2007, 316, 970–972. [Google Scholar] [CrossRef] [PubMed]
- Goblirsch, M. Nosema ceranae disease of the honey bee (Apis mellifera). Apidologie 2017, 49, 131–150. [Google Scholar] [CrossRef]
- Solomon, K.R.; Williams, W.M.; Mackay, D.; Purdy, J.; Giddings, J.M.; Giesy, J.P. Properties and uses of chlorpyrifos in the United States. Rev. Environ. Contam. Toxicol. 2014, 231, 13–34. [Google Scholar] [CrossRef]
- Stout, J.C.; Morales, C.L. Ecological impacts of invasive alien species on bees. Apidologie 2009, 40, 388–409. [Google Scholar] [CrossRef]
- Jensen, A.B.; Palmer, K.A.; Boomsma, J.J.; Pedersen, B.V. Varying degrees of Apis mellifera ligustica introgression in protected populations of the black honeybee, Apis mellifera mellifera, in northwest Europe. Mol. Ecol. 2004, 14, 93–106. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Lee, M.L.; Yunusbaev, U.; Nikolenko, A.; Kwon, H.W. Estimation of C-derived introgression into A. m. mellifera colonies in the Russian Urals using microsatellite genotyping. Genes Genom. 2020, 42, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.B.; Pedersen, B.V. Honeybee Conservation: A case story from Læsø island, Denmark. In Beekeeping and Conserving Biodiversity of Honeybee. Sustainable Bee Breeding. Theoretical and Practical Guide; Lodesani, M., Costa, C., Eds.; Northern Bee Books: Hebden Bridge, UK, 2005; pp. 142–164. [Google Scholar]
- Strange, J.P.; Garnery, L.; Sheppard, W.S. Morphological and molecular characterization of the Landes honey bee (Apis mellifera L.) ecotype for genetic conservation. J. Insect Conserv. 2008, 12, 527–537. [Google Scholar] [CrossRef]
- Bieńkowska, M.; Splitt, A.; Wegrzynowicz, P.; Maciorowski, R. The Buzz Changes within Time: Native Apis mellifera mellifera Honeybee Subspecies Less and Less Popular among Polish Beekeepers Since 1980. Agriculture 2021, 11, 652. [Google Scholar] [CrossRef]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.-B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef]
- The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbil, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Crozier, R.H.; Crozier, Y.C. The mitochondrial genome of the honeybee Apis mellifera: Complete sequence and genome organization. Genetics 1993, 133, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Meusel, M.S.; Moritz, R.F.A. Transfer of Paternal Mitochondrial DNA during Fertilization of Honeybee (Apis mellifera L.) Eggs. Curr. Genet. 1993, 24, 539–543. [Google Scholar] [CrossRef]
- Ilyasov, R.; Nikolenko, A.; Tuktarov, V.; Goto, K.; Takahashi, J.I.; Kwon, H.W. Comparative Analysis of Mitochondrial Genomes of the Honey Bee Subspecies A. m. caucasica and A. m. carpathica and Refinement of Their Evolutionary Lineages. J. Apic. Res. 2019, 58, 567–579. [Google Scholar] [CrossRef]
- Haddad, N.J. Mitochondrial genome of the Levant Region honeybee, Apis mellifera syriaca (Hymenoptera: Apidae). Mitochondrial DNA Part A 2015, 27, 4067–4068. [Google Scholar] [CrossRef] [PubMed]
- Garnery, L.; Cornuet, J.M.; Solignac, M. Evolutionary history of the honey bee Apis mellifera inferred from mitochondrial DNA analysis. Mol. Ecol. 1992, 1, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.M. Molecular confirmation of a fourth lineage in honeybees from the Near East. Apidologie 2000, 31, 167–180. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Loiseau, A.; Oldroyd, B.P.; Hepburn, H.R.; Solignac, M.; Cornuet, J.M. Genetic diversity of the honeybee in Africa: Microsatellite and mitochondrial data. Heredity 2001, 86, 420. [Google Scholar] [CrossRef]
- Alburaki, M.; Bertrand, B.; Legout, H.; Moulin, S.; Alburaki, A.; Sheppard, W.; Sheppard, W.S.; Garnery, L. A Fifth Major Genetic Group among Honeybees Revealed in Syria. BMC Genet. 2013, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Tihelka, E.; Cai, C.; Pisani, D.; Donoghue, P.C.J. Mitochondrial genomes illuminate the evolutionary history of the Western honey bee (Apis mellifera). Sci. Rep. 2020, 10, 14515. [Google Scholar] [CrossRef]
- Moškrič, A.; Marinč, A.; Ferk, P.; Leskošek, B.; Mosbech, M.B.; Bunikis, I.; Pettersson, O.V.; Soler, L.; Prešern, J. The Carniolan Honeybee from Slovenia—A Complete and Annotated Mitochondrial Genome with Comparisons to Closely Related Apis mellifera Subspecies. Insects 2022, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Guichard, M.; Dainat, B.; Eynard, S.; Vignal, A.; Servin, B.; Beestrong Consortium; Neuditschko, M. Identification of quantitative trait loci associated with calmness and gentleness in honey bees using whole-genome sequences. Anim. Genet. 2021, 52, 472–481. [Google Scholar] [CrossRef]
- Ruttner, F. Naturgeschichte der Honigbienen; Ehrenwirt Verlag: München, Germany, 1992; p. 350. [Google Scholar]
- Muñoz, I.; Dall’Olio, R.; Lodesani, M.; De la Rúa, P. Population genetic structure of coastal Croatian honeybees (Apis mellifera carnica). Apidologie 2009, 40, 617–626. [Google Scholar] [CrossRef]
- Muñoz, I.; De la Rúa, P. Wide genetic diversity in Old World honey bees threaten by introgression. Apidologie 2021, 52, 200–217. [Google Scholar] [CrossRef]
- Ludányi, I. A himalájai méh és az Apis mellifera alfajai. Méhészet 1997, 45, 10–11. [Google Scholar]
- Performance Testing Codex of the Pannonian Bee (Magyar Méhtenyésztők Országos Egyesülete: Pannon Méh Teljesítményvizsgálati Kódex 2019). Available online: https://portal.nebih.gov.hu/documents/10182/43858/Pannon+m%C3%A9h+teljes%C3%ADtm%C3%A9nyvizsg%C3%A1lati+k%C3%B3dex+2019.pdf/dbee9a9b-0a6f-f454-04ee-31b962925d1d (accessed on 6 May 2019).
- Hungarian Bee-Breeders’ Association (HBBA): Introduction of the Breeder Organisation (Tenyésztőszervezet Bemutatkozása). Available online: https://mmoe.hu/ (accessed on 12 April 2019).
- Péntek-Zakar, E.; Oleksa, A.; Borowik, T.; Kusza, S. Population structure of honey bees in the Carpathian Basin (Hungary) confirms introgression from surrounding subspecies. Ecol. Evol. 2015, 5, 5456–5467. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Moya, A.; Ayala, F.J. Evolution of mitochondrial DNA in Drosophila subobscura. Proc. Natl. Acad. Sci. USA 1986, 83, 8649–8653. [Google Scholar] [CrossRef] [PubMed]
- Péntek-Zakar, E. A Magyarországi Mézelőméh-Populációkban (APIS MELLIFERA CARNICA PANNONICA POLL.) Megjelenő Határ Menti Fajták Kutatása Genetikai és Morfológiai Módszerekkel. Doctoral Dissertation, University of Debrecen, Debrecen, Hungary, 2014. [Google Scholar]
- Magnus, R.M.; Tripodi, A.D.; Szalanski, A.L. Mitochondrial DNA Diversity of Honey Bees (Apis mellifera) from Unmanaged Colonies and Swarms in the United States. Biochem. Genet. 2014, 52, 245–257. [Google Scholar] [CrossRef]
- Estoup, A.; Garnery, L.; Solignac, M.; Cornuet, J.M. Microsatellite variation in honey bee (Apis mellifera L.) populations: Hierarchical genetic structure and test of the infinite allele and stepwise mutation models. Genetics 1995, 140, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Solignac, M.; Vautrin, D.; Loiseau, A.; Mougel, F.; Baudry, E.; Estoup, A.; Garnery, L.; Haberl, M.; Cornuet, J.M. Five hundred and fifty microsatellite markers for the study of the honeybee (Apis mellifera L.) genome. Mol. Ecol. Notes 2003, 3, 307–311. [Google Scholar] [CrossRef]
- Garnery, L.; Franck, P.; Baudry, E.; Vautrin, D.; Cornuet, J.M.; Solignac, M. Genetic biodiversity of the west European honeybee (Apis mellifera mellifera and Apis mellifera iberica) II. Microsatellite DNA. Genet. Sel. Evol. 1998, 30, 49–74. [Google Scholar] [CrossRef]
- Franck, P.; Garnery, L.; Solignac, M.; Cornuet, J.M. The origin of West European subspecies of honeybees (Apis mellifera): New insights from microsatellite and mitochondrial data. Evolution 1998, 52, 1119–1134. [Google Scholar] [CrossRef] [PubMed]
- Techer, M.A.; Clémencet, J.; Turpin, P.; Volbert, N.; Reynaud, B.; Delatte, H. Genetic characterization of the honeybee (Apis mellifera) population of Rodrigues Island, based on microsatellite and mitochondrial DNA. Apidologie 2015, 46, 445–454. [Google Scholar] [CrossRef]
- Blacket, M.J.; Robin, C.; Good, R.T.; Lee, S.F.; Miller, A.D. Universal primers for fluorescent labelling of PCR fragments-an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 2012, 12, 456–463. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.G.; Sharp, P.M. CLUSTAL: A package for performing multiple sequence alignment on a microcomputer. Gene 1988, 73, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Altschul, F.S.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Park, S.D.E. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Doctoral Dissertation, University of Dublin, Dublin, Ireland, 2001. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Sušnik, S.; Kozmus, P.; Poklukar, J.; Megli, V. Molecular characterisation of indigenous Apis mellifera carnica in Slovenia. Apidologie 2004, 35, 623–636. [Google Scholar] [CrossRef]
- Alburaki, M.; Madella, S.; Lopez, J.; Bouga, M.; Chen, Y.; vanEngelsdorp, D. Honey bee populations of the USA display restrictions in their mtDNA haplotype diversity. Front. Genet. 2023, 13, 1092121. [Google Scholar] [CrossRef] [PubMed]
- Patenković, A.; Tanasković, M.; Erić, P.; Erić, K.; Mihajlović, M.; Stanisavljević, L.; Davidović, S. Urban ecosystem drives genetic diversity in feral honey bee. Sci. Rep. 2022, 12, 17692. [Google Scholar] [CrossRef] [PubMed]
- Coroian, C.O.; Muñoz, I.; Schlüns, E.A.; Paniti-Teleky, O.R.; Erler, S.; Furdui, E.M.; Mărghitaş, L.A.; Dezmirean, S.S.; Schlüns, H.; De La Rúa, P.; et al. Climate rather than geography separates two European honeybee subspecies. Mol. Ecol. 2014, 23, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Tanasković, M.; Erić, P.; Patenković, A.; Erić, K.; Mihajlović, M.; Tanasić, V.; Stanisavljević, L.; Davidović, S. MtDNA Analysis Indicates Human-Induced Temporal Changes of Serbian Honey Bees Diversity. Insects 2021, 12, 767. [Google Scholar] [CrossRef]
- Yin, L.; Ji, T.; Chen, G.; Peng, W. Genetic characterization of three breeds of high royal jelly producing honeybee (Apis mellifera ligustica) in China. Afr. J. Agric. Res. 2011, 6, 331–337. [Google Scholar]
- Ilyasov, R.A.; Poskryakov, A.V.; Petukhov, A.V.; Nikolenko, A.G. Molecular genetic analysis of five extant reserves of black honeybee Apis melifera melifera in the Urals and the Volga region. Russ. J. Genet. 2016, 52, 828–839. [Google Scholar] [CrossRef]
- Balázs, R.; Edviné Meleg, E.; Hidas, A.; Rácz, T.; Zajácz, E.; Pálinkás-Bodzsár, N. Pannonméh-állományok genetikai diverzitásának vizsgálata molekuláris markerekkel. Méhészet 2023, 71, 22–24. (In Hungarian) [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplotype | Number of Individuals | Position of the Polymorphism (bp) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | MF | NMF | REF | KRAJ | 32 | 143 | 170 | 171 | 270 | 293 | 370 | 376 | 412 | 573 | |
| H1 | 106 | 54 | 19 | 24 | 9 | T | C | A | G | T | T | C | A | C | T |
| H2 | 7 | 7 | . | A | . | . | . | C | . | . | . | A | |||
| H3 | 1 | 1 | . | . | G | . | . | . | . | . | . | . | |||
| H4 | 2 | 2 | . | . | . | A | . | . | . | . | . | . | |||
| H5 | 17 | 4 | 13 | . | . | . | . | . | . | T | . | . | . | ||
| H6 | 8 | 8 | C | . | . | . | C | . | . | G | T | C | |||
| Group | H | Haplotypes | Hd ± SD | π ± SD | Fs | D | p (D*) |
|---|---|---|---|---|---|---|---|
| MF | 4 | H1, H3, H4, H5 | 0.214 ± 0.068 | 0.00032 ± 0.00011 | −2.610 | −1.29295 | ns |
| NMF | 2 | H1, H5 | 0.498 ± 0.039 | 0.00072 ± 0.00006 | 1.670 | 1.53512 | ns |
| REF | 2 | H1, H6 | 0.387 ± 0.078 | 0.00278 ± 0.00056 | 6.099 | 1.50012 | ns |
| KRAJ | 2 | H1, H2 | 0.525 ± 0.055 | 0.00226 ± 0.00024 | 4.038 | 2.15124 | ** |
| TOTAL | 6 | H1, H2, H3, H4, H5, H6 | 0.417 ± 0.049 | 0.00154 ± 0.00027 | 0.147 | −1.14724 | ns |
| Haplotype | Number of Individuals | Position of the Polymorphism (bp) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | MF | NMF | REF | KRAJ | 17 | 61 | 113 | 134 | 179 | 314 | |
| HE1 (C1) | 45 | 24 | 13 | 7 | 1 | . | . | . | . | T | C |
| HE2 (C2p) | 13 | 1 | 12 | . | A | . | . | . | . | ||
| HE3 (C2i) | 2 | 2 | . | . | . | A | . | . | |||
| HE4 (C2d) | 41 | 20 | 14 | 7 | T | T | A | G | C | T | |
| HE5 (C2e) | 22 | 9 | 5 | 8 | - | . | . | . | . | . | |
| HE6 (C2l) | 1 | 1 | . | . | - | . | . | . | |||
| Group | Sample Size | MNA ± SD | HE ± SD | HO ± SD | FIS | HW |
|---|---|---|---|---|---|---|
| MF | 64 | 9.3 ± 4.93 | 0.59 ± 0.046 | 0.54 ± 0.014 | 0.088 | *** |
| NMF | 32 | 6.5 ± 3.27 | 0.55 ± 0.052 | 0.52 ± 0.020 | 0.053 | * |
| REF | 32 | 7.2 ± 3.51 | 0.64 ± 0.051 | 0.62 ± 0.019 | 0.029 | ns |
| KRAJ | 16 | 3.7 ± 2.18 | 0.47 ± 0.054 | 0.47 ± 0.028 | 0.013 | ns |
| MF | NMF | REF | KRAJ | |
|---|---|---|---|---|
| MF | 0.005 | 0.000 | 0.000 | |
| NMF | 0.008 | 0.000 | 0.000 | |
| REF | 0.042 | 0.047 | 0.000 | |
| KRAJ | 0.032 | 0.037 | 0.071 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balazs, R.; Molnar, T.G.; Edvine Meleg, E.; Hidas, A.; Zajacz, E.; Racz, T.; Palinkas-Bodzsar, N. Evolutionary Origin and Genetic Diversity of the Pannonian Ecotype of Apis mellifera carnica Colonies in Hungary Based on Mitochondrial DNA and Microsatellite Markers. Biology 2025, 14, 475. https://doi.org/10.3390/biology14050475
Balazs R, Molnar TG, Edvine Meleg E, Hidas A, Zajacz E, Racz T, Palinkas-Bodzsar N. Evolutionary Origin and Genetic Diversity of the Pannonian Ecotype of Apis mellifera carnica Colonies in Hungary Based on Mitochondrial DNA and Microsatellite Markers. Biology. 2025; 14(5):475. https://doi.org/10.3390/biology14050475
Chicago/Turabian StyleBalazs, Reka, Tamas Gergely Molnar, Erika Edvine Meleg, Andras Hidas, Edit Zajacz, Timea Racz, and Nora Palinkas-Bodzsar. 2025. "Evolutionary Origin and Genetic Diversity of the Pannonian Ecotype of Apis mellifera carnica Colonies in Hungary Based on Mitochondrial DNA and Microsatellite Markers" Biology 14, no. 5: 475. https://doi.org/10.3390/biology14050475
APA StyleBalazs, R., Molnar, T. G., Edvine Meleg, E., Hidas, A., Zajacz, E., Racz, T., & Palinkas-Bodzsar, N. (2025). Evolutionary Origin and Genetic Diversity of the Pannonian Ecotype of Apis mellifera carnica Colonies in Hungary Based on Mitochondrial DNA and Microsatellite Markers. Biology, 14(5), 475. https://doi.org/10.3390/biology14050475