The Nuclear Envelope in Lipid Metabolism and Pathogenesis of NAFLD

Abstract
:Simple Summary
Abstract
1. Introduction
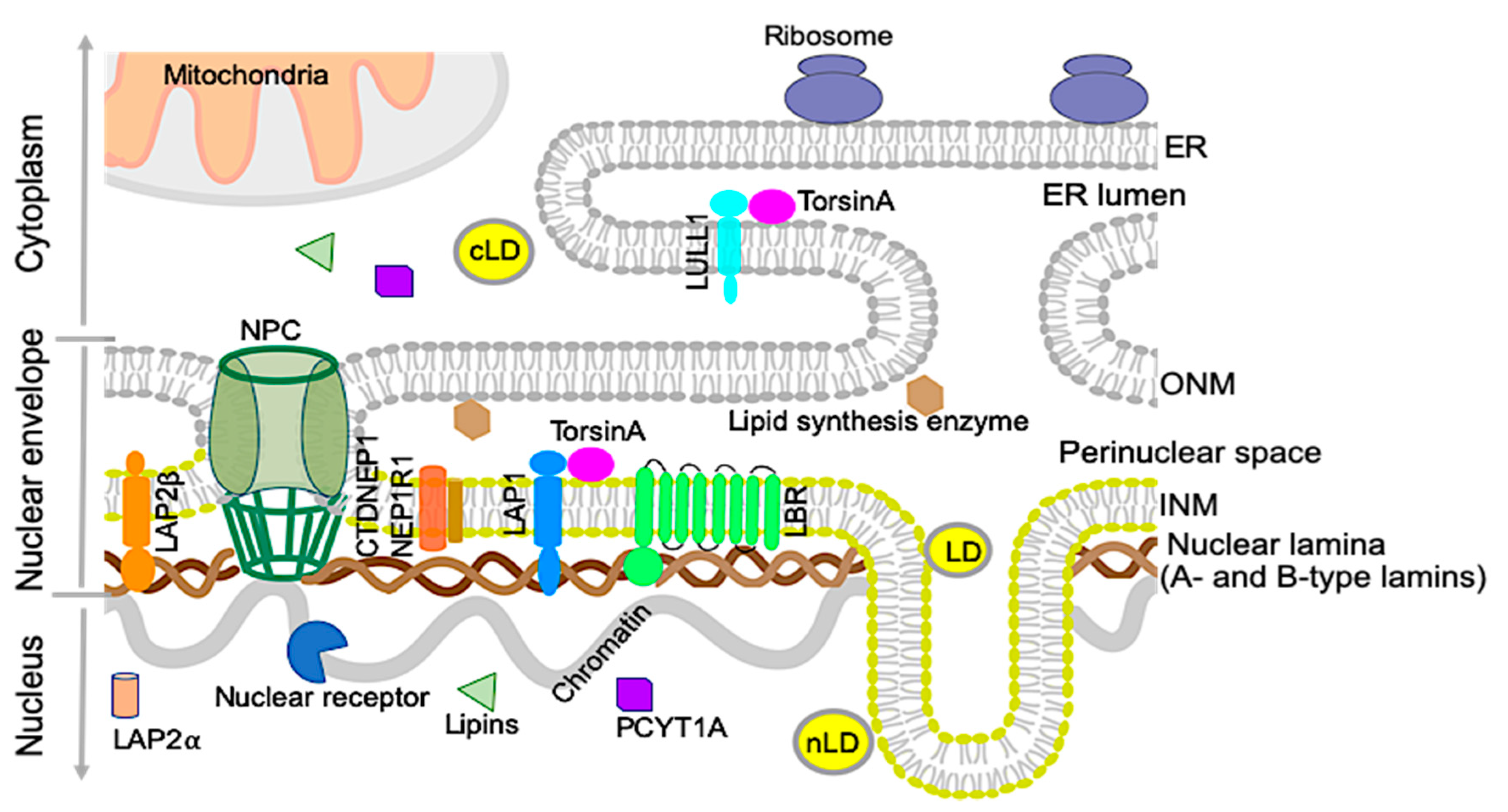
2. Nuclear Envelope and Lipid Metabolism
2.1. Nuclear Lipid Droplets
2.2. Nuclear Lamins and Membrane Proteins Implicated in Lipid Metabolism
2.2.1. Lamin B receptor (LBR)
2.2.2. Lipins and CTDNEP1/NEP1R1
2.2.3. Choline-Phosphate Cytidylyltransferase A (PCYT1A)
2.2.4. A-Type Lamins
2.2.5. LAP2
2.2.6. Lamin B1
3. The TorsinA/LAP1 Complex in Lipid Metabolism and NASH Development
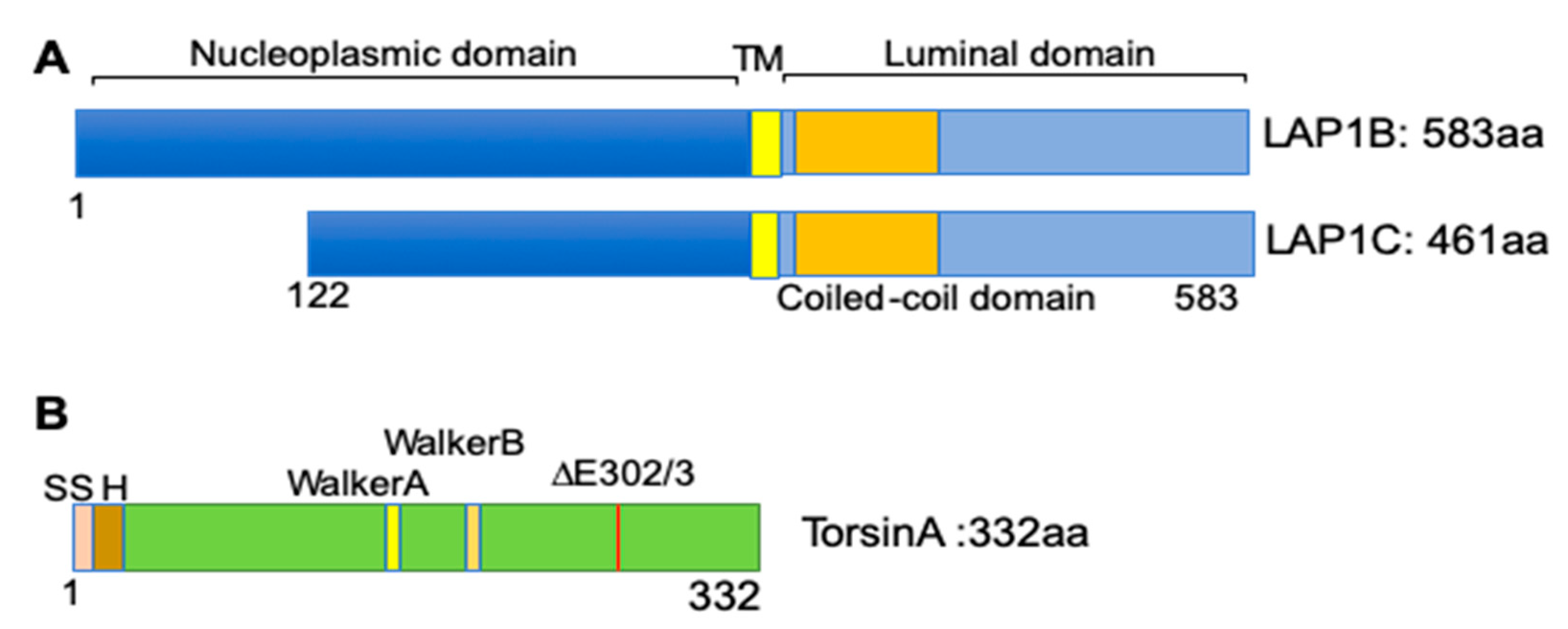
3.1. The Discovery of LAP1 and TorsinA Interaction and Its Implications in Human Diseases
3.2. The TorsinA/LAP1 Complex in LD Biogenesis and Hepatic Lipid Secretion
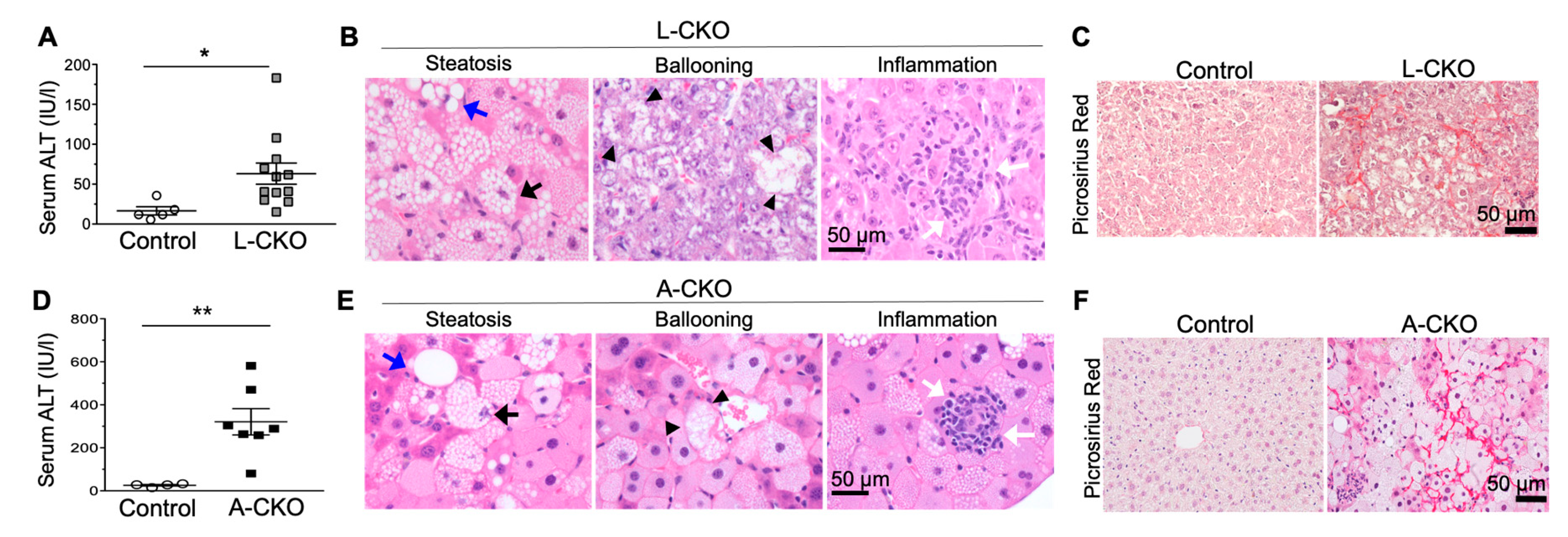
3.3. NASH Development in Chow-Fed Mice with Depletion of LAP1 or TorsinA
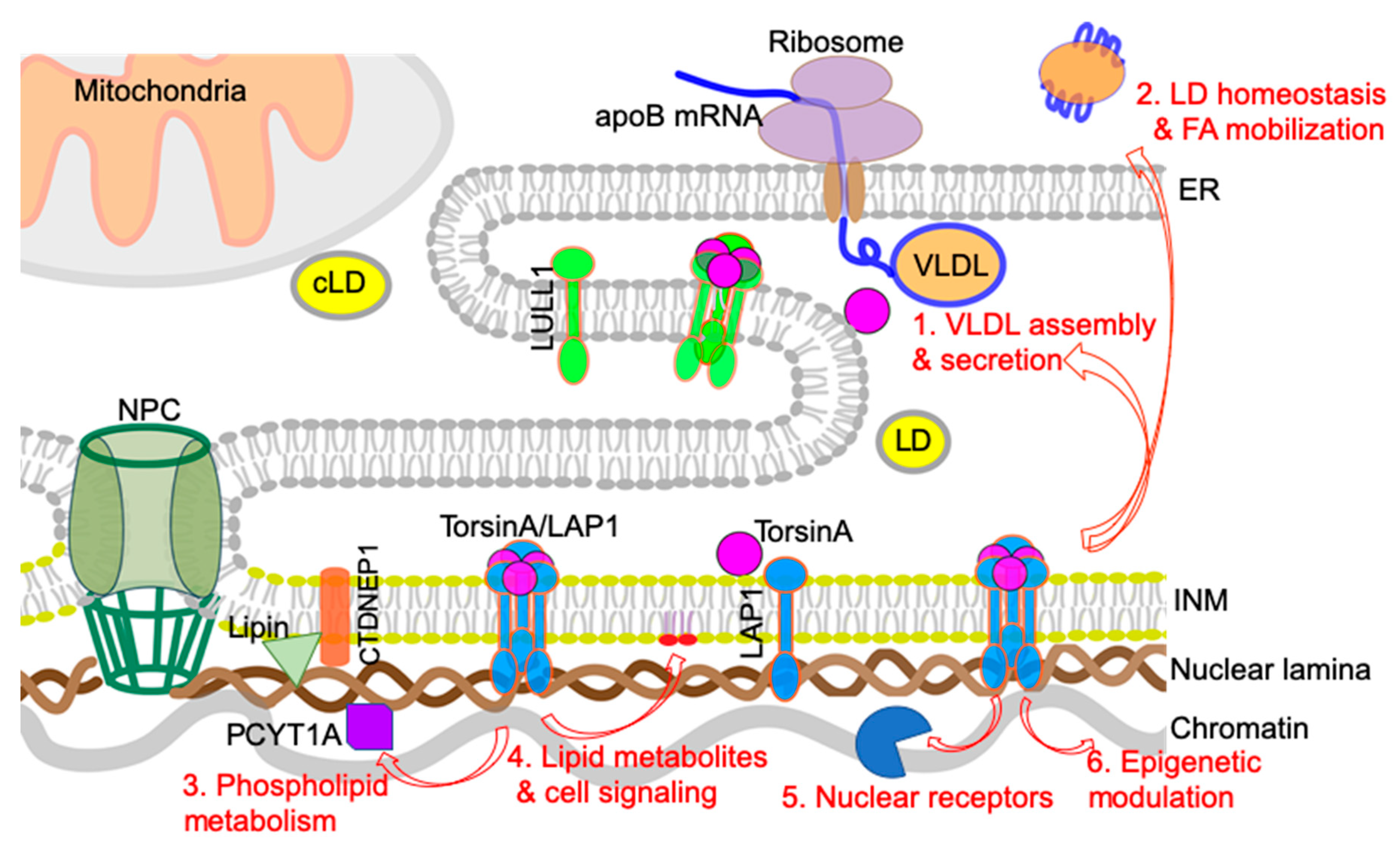
4. Possible Cellular Mechanisms Connecting Defective Nuclear Envelope Proteins to NAFLD/NASH Pathogenesis
4.1. VLDL Assembly and Secretion
4.2. LD Homeostasis and Fatty Acid (FA) Mobilization/Oxidation
4.3. Phospholipid Metabolism: Balance between Lipid Storage and Membrane Lipid Synthesis
4.4. Lipid Metabolites and Lipid-Mediated Cell Signaling
4.5. Nuclear Receptors and Transcription Factors
4.6. Epigenetic Regulation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFL development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. 2013, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Loomba, R.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; Ratziu, V. Current and future therapeutic regimens for nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2018, 68, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.-S. Pathogenesis of nonalcoholic steatohepatitis and hormone-based therapeutic approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Barbara, M.; Scott, A.; Alkhouri, N. New insights into genetic predisposition and novel therapeutic targets for nonalcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2018, 7, 372–381. [Google Scholar] [CrossRef]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The pathogenesis of nonalcoholic fatty liver disease: Interplay between diet, gut microbiota, and genetic background. Gastroenterol. Res. Pract. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206. [Google Scholar] [CrossRef]
- Connolly, J.J.; Ooka, K.; Lim, J.K. Future pharmacotherapy for non-alcoholic steatohepatitis (NASH): Review of phase 2 and 3 trials. J. Clin. Transl. Hepatol. 2018, 6, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.T.; Worman, H.J. The nuclear envelope as a signaling node in development and disease. Dev. Cell 2009, 17, 626–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worman, H.J.; Schirmer, E.C. Nuclear membrane diversity: Underlying tissue-specific pathologies in disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Östlund, C.; Chang, W.; Gundersen, G.G.; Worman, H.J. Pathogenic mutations in genes encoding nuclear envelope proteins and defective nucleocytoplasmic connections. Exp. Biol. Med. 2019, 244, 1333–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelz, A.; Debler, E.W.; Blobel, G. The structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef]
- Siniossoglou, S. Lipins, lipids and nuclear envelope structure. Traffic 2009, 10, 1181–1187. [Google Scholar] [CrossRef]
- Jacquemyn, J.; Cascalho, A.; Goodchild, R.E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 2017, 18, 1905–1921. [Google Scholar] [CrossRef]
- Sołtysik, K.; Ohsaki, Y.; Fujimoto, T. Duo in a Mystical Realm—Nuclear Lipid Droplets and the Inner Nuclear Membrane. Contact 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Sołtysik, K.; Ohsaki, Y.; Tatematsu, T.; Cheng, J.; Fujimoto, T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Schlieker, C. Lipid and protein dynamics that shape nuclear envelope identity. Mol. Biol. Cell 2020, 31, 1315–1323. [Google Scholar] [CrossRef]
- Lagrutta, L.C.; Montero-Villegas, S.; Layerenza, J.P.; Sisti, M.S.; García de Bravo, M.M.; Ves-Losada, A. Reversible nuclear-lipid-droplet morphology induced by oleic acid: A link to cellular-lipid metabolism. PLoS ONE 2017, 12, e0170608. [Google Scholar] [CrossRef]
- Layerenza, J.P.; González, P.; De Bravo, M.G.; Polo, M.P.; Sisti, M.S.; Ves-Losada, A. Nuclear lipid droplets: A novel nuclear domain. BBA-Mol. Cell Biol. Lipids 2013, 1831, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V., Jr.; Walther, T.C. Lipid droplets go nuclear. J. Cell Biol. 2016, 212, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Hernandez-Ono, A.; Fedotova, T.; Ostlund, C.; Lee, M.J.; Gibeley, S.B.; Liang, C.C.; Dauer, W.T.; Ginsberg, H.N.; Worman, H.J. Nuclear envelope-localized torsinA-LAP1 complex regulates hepatic VLDL secretion and steatosis. J. Clin. Investig. 2019, 130, 4885–4900. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Kawai, T.; Yoshikawa, Y.; Cheng, J.; Jokitalo, E.; Fujimoto, T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 2016, 212, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanauska, A.; Köhler, A. The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 2018, 174, 700–715.e18. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Yuan, J.; Blobel, G.; Georgatos, S.D. A lamin B receptor in the nuclear envelope. Proc. Natl. Acad. Sci. USA 1988, 85, 8531–8534. [Google Scholar] [CrossRef] [Green Version]
- Holmer, L.; Pezhman, A.; Worman, H.J. The human lamin B receptor/sterol reductase multigene family. Genomics 1998, 54, 469–476. [Google Scholar] [CrossRef]
- Worman, H.J.; Evans, C.D.; Blobel, G. The lamin B receptor of the nuclear envelope inner membrane: A polytopic protein with eight potential transmembrane domains. J. Cell Biol. 1990, 111, 1535–1542. [Google Scholar] [CrossRef]
- Hoffmann, K.; Dreger, C.K.; Olins, A.L.; Olins, D.E.; Shultz, L.D.; Lucke, B.; Karl, H.; Kaps, R.; Müller, D.; Vayá, A. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger–Huet anomaly). Nat. Genet. 2002, 31, 410–414. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; Mooyer, P.; Van Noort, G.; Kelley, R.I.; Wilcox, W.R.; Wanders, J.R.; Hennekam, C.R.; Oosterwijk, C.J. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3β-hydroxysterol Δ14-reductase deficiency due to mutations in the lamin B receptor gene. Am. J. Hum. Genet. 2003, 72, 1013–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, P.; Fischer, B.; Mann, A.; Mansour, S.; Rossier, E.; Veen, M.; Lang, C.; Baasanjav, S.; Kieslich, M.; Brossuleit, K. Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus 2010, 1, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, P.-L.; Zhao, C.; Turner, E.; Schlieker, C. The Lamin B receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. eLife 2016, 5, e16011. [Google Scholar] [CrossRef] [PubMed]
- Reue, K.; Dwyer, J.R. Lipin proteins and metabolic homeostasis. J. Lipid Res. 2009, 50, S109–S114. [Google Scholar] [CrossRef] [Green Version]
- Siniossoglou, S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. BBA-Mol. Cell Biol. Lipids 2013, 1831, 575–581. [Google Scholar] [CrossRef]
- Kim, Y.; Gentry, M.S.; Harris, T.E.; Wiley, S.E.; Lawrence, J.C.; Dixon, J.E. A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6596–6601. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Bahmanyar, S.; Zhang, P.; Grishin, N.; Oegema, K.; Crooke, R.; Graham, M.; Reue, K.; Dixon, J.E.; Goodman, J.M. Nuclear envelope phosphatase 1-regulatory subunit 1 (formerly TMEM188) is the metazoan Spo7p ortholog and functions in the lipin activation pathway. J. Biol. Chem. 2012, 287, 3123–3137. [Google Scholar] [CrossRef] [Green Version]
- Jacquemyn, J.; Foroozandeh, J.; Vints, K.; Swerts, J.; Verstreken, P.; Gounko, N.V.; Gallego, S.F.; Goodchild, R. The Torsin/NEP1R1-CTDNEP1/Lipin axis regulates nuclear envelope lipid metabolism for nuclear pore complex insertion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Aitchison, A.J.; Arsenault, D.J.; Ridgway, N.D. Nuclear-localized CTP: Phosphocholine cytidylyltransferase α regulates phosphatidylcholine synthesis required for lipid droplet biogenesis. Mol. Biol. Cell 2015, 26, 2927–2938. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Hegele, R.A. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speckman, R.A.; Garg, A.; Du, F.; Bennett, L.; Veile, R.; Arioglu, E.; Taylor, S.I.; Lovett, M.; Bowcock, A.M. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet. 2000, 66, 1192–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegele, R.A. Familial partial lipodystrophy: A monogenic form of the insulin resistance syndrome. Mol. Genet. Metab. 2000, 71, 539–544. [Google Scholar] [CrossRef]
- Lüdtke, A.; Genschel, J.; Brabant, G.; Bauditz, J.; Taupitz, M.; Koch, M.; Wermke, W.; Worman, H.J.; Schmidt, H.H.-J. Hepatic steatosis in Dunnigan-type familial partial lipodystrophy. Am. J. Gastroenterol. 2005, 100, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Guenantin, A.; Briand, N.; Bidault, G.; Afonso, P.; Bereziat, V.; Vatier, C.; Lascols, O.; Caron-Debarle, M.; Capeau, J.; Vigouroux, C. Nuclear envelope-related lipodystrophies. Semin. Cell Dev. Biol. 2014, 29, 148–157. [Google Scholar] [CrossRef]
- Kwan, R.; Brady, G.F.; Brzozowski, M.; Weerasinghe, S.V.; Martin, H.; Park, M.J.; Brunt, M.J.; Menon, R.K.; Tong, X.; Yin, L.; et al. Hepatocyte-Specific Deletion of Mouse Lamin A/C Leads to Male-Selective Steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 365–383. [Google Scholar] [CrossRef] [Green Version]
- Brady, G.F.; Kwan, R.; Ulintz, P.J.; Nguyen, P.; Bassirian, S.; Basrur, V.; Nesvizhskii, A.I.; Loomba, R.; Omary, M.B. Nuclear lamina genetic variants, including a truncated LAP2, in twins and siblings with nonalcoholic fatty liver disease. Hepatology 2017, 67, 1710–1725. [Google Scholar] [CrossRef] [Green Version]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptacek, L.J.; Fu, Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Nmezi, B.; Giorgio, E.; Raininko, R.; Lehman, A.; Spielmann, M.; Koenig, M.K.; Adejumo, R.; Knight, M.; Gavrilova, R.; Alturkustani, M.; et al. Genomic deletions upstream of lamin B1 lead to atypical autosomal dominant leukodystrophy. Neurol. Genet. 2019, 5, e305. [Google Scholar] [CrossRef] [Green Version]
- Padiath, Q.S. Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina. Front. Cell Dev. Biol. 2019, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolyan, H.; Tyurina, Y.Y.; Hernandez, M.; Amoscato, A.A.; Sparvero, L.J.; Nmezi, B.C.; Lu, Y.; Estecio, M.R.; Lin, K.; Chen, J.; et al. Defects of Lipid Synthesis Are Linked to the Age-Dependent Demyelination Caused by Lamin B1 Overexpression. J. Neurosci. 2015, 35, 12002–12017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, A.; Gerace, L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J. Cell Biol. 1988, 107, 2029–2036. [Google Scholar] [CrossRef]
- Foisner, R.; Gerace, L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 1993, 73, 1267–1279. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Méndez-López, I.; Hong, M.; Wang, Y.; Tanji, K.; Wu, W.; Shugol, L.; Krauss, R.S.; Dauer, W.T.; Worman, H.J. Lamina-associated polypeptide 1 is dispensable for embryonic myogenesis but required for postnatal skeletal muscle growth. Hum. Mol. Genet. 2017, 26, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Domingues, S.C.; Costa, P.; Muller, T.; Galozzi, S.; Marcus, K.; e Silva, E.F.d.C.; e Silva, O.A.d.C.; Rebelo, S. Identification of a novel human LAP1 isoform that is regulated by protein phosphorylation. PLoS ONE 2014, 9, e113732. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Dauer, W.T.; Worman, H.J. Lamina-associated polypeptide 1: Protein interactions and tissue-selective functions. Semin. Cell Dev. Biol. 2014, 29C, 164–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Mendez-Lopez, I.; Wang, Y.; Hays, A.P.; Tanji, K.; Lefkowitch, J.H.; Schulze, P.C.; Worman, H.J.; Dauer, W.T. Lamina-Associated Polypeptide-1 Interacts with the Muscular Dystrophy Protein Emerin and Is Essential for Skeletal Muscle Maintenance. Dev. Cell 2013, 26, 591–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Rebelo, S.; Van Kleeff, P.J.; Kim, C.E.; Dauer, W.T.; Fardilha, M.; da Cruz e Silva, O.A.; da Cruz e Silva, E.F. The nuclear envelope protein, LAP1B, is a novel protein phosphatase 1 substrate. PLoS ONE 2013, 8, e76788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, J.B.; Da Cruz e Silva, O.A.; Rebelo, S. Lamina associated polypeptide 1 (LAP1) interactome and its functional features. Membranes 2016, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.D.; Martins, F.; Santos, M.; Müeller, T.; da Cruz e Silva, O.A.; Rebelo, S. Nuclear Accumulation of LAP1: TRF2 Complex during DNA Damage Response Uncovers a Novel Role for LAP1. Cells 2020, 9, 1804. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, R.E.; Dauer, W.T. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 2005, 168, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; De Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rampello, A.J.; Prophet, S.M.; Schlieker, C. The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease. Biomolecules 2020, 10, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsinA mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical AAA+ ATPases. Crit. Rev. Biochem. Mol. 2015, 50, 532–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of Torsin ATPases by LAP1 and LULL1. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554. [Google Scholar] [CrossRef] [Green Version]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (LAP1) activates Torsin. eLife 2014, 3, e03239. [Google Scholar] [CrossRef]
- Demircioglu, F.E.; Sosa, B.A.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. eLife 2016, 5, e17983. [Google Scholar] [CrossRef]
- Kayman-Kurekci, G.; Talim, B.; Korkusuz, P.; Sayar, N.; Sarioglu, T.; Oncel, I.; Sharafi, P.; Gundesli, H.; Balci-Hayta, B.; Purali, N.; et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: A novel gene related to nuclear envelopathies. Neuromuscul. Disord. 2014, 24, 624–633. [Google Scholar] [CrossRef]
- Dorboz, I.; Coutelier, M.; Bertrand, A.T.; Caberg, J.H.; Elmaleh-Berges, M.; Laine, J.; Stevanin, G.; Bonne, G.; Boespflug-Tanguy, O.; Servais, L. Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1. Orphanet. J. Rare Dis. 2014, 9, 174. [Google Scholar] [CrossRef] [Green Version]
- Ghaoui, R.; Benavides, T.; Lek, M.; Waddell, L.B.; Kaur, S.; North, K.N.; MacArthur, D.G.; Clarke, N.F.; Cooper, S.T. TOR1AIP1 as a cause of cardiac failure and recessive limb-girdle muscular dystrophy. Neuromuscul. Disord. 2016, 26, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Fichtman, B.; Zagairy, F.; Biran, N.; Barsheshet, Y.; Chervinsky, E.; Neriah, Z.B.; Shaag, A.; Assa, M.; Elpeleg, O.; Harel, A. Combined loss of LAP1B and LAP1C results in an early onset multisystemic nuclear envelopathy. Nat. Commun. 2019, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Lessel, I.; Chen, M.-J.; Lüttgen, S.; Arndt, F.; Fuchs, S.; Meien, S.; Thiele, H.; Jones, J.R.; Shaw, B.R.; Crossman, D.K. Two novel cases further expand the phenotype of tor1aip1-associated nuclear envelopathies. Hum. Genet. 2020, 139, 483–498. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Le Dour, C.; Sera, F.; Iwata, S.; Homma, S.; Joseph, L.C.; Morrow, J.P.; Dauer, W.T.; Worman, H.J. Depletion of lamina-associated polypeptide 1 from cardiomyocytes causes cardiac dysfunction in mice. Nucleus 2014, 5, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.E.; Perez, A.; Perkins, G.; Ellisman, M.H.; Dauer, W.T. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9861–9866. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, H.N.; Fisher, E.A. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res. 2009, 50, S162–S166. [Google Scholar] [CrossRef] [Green Version]
- Teng, B.; Burant, C.F.; Davidson, N.O. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science 1993, 260, 1816–1819. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Smagris, E.; Gilyard, S.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J. Biol. Chem. 2016, 291, 10659–10676. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, M.; Moulin, P.; Roy, P.; Samson-Bouma, M.E.; Collardeau-Frachon, S.; Chebel-Dumont, S.; Peretti, N.; Dumortier, J.; Zoulim, F.; Fontanges, T. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J. Hepatol. 2014, 61, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.-J.; Lee, M.-Y.; Wang, Y.-T.; Jiang, H.-J.; Lin, P.-C.; Yang, Y.-H.C.; Kuo, K.-K. MTTP-297H polymorphism reduced serum cholesterol but increased risk of non-alcoholic fatty liver disease-a cross-sectional study. BMC Med. Genet. 2015, 16, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pariyarath, R.; Wang, H.; Aitchison, J.D.; Ginsberg, H.N.; Welch, W.J.; Johnson, A.E.; Fisher, E.A. Co-translational interactions of apoprotein B with the ribosome and translocon during lipoprotein assembly or targeting to the proteasome. J. Biol. Chem. 2001, 276, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Park, S.; Kodali, V.K.; Han, J.; Yip, T.; Chen, Z.; Davidson, N.O.; Kaufman, R.J. Identification of protein disulfide isomerase 1 as a key isomerase for disulfide bond formation in apolipoprotein B100. Mol. Biol. Cell 2015, 26, 594–604. [Google Scholar] [CrossRef]
- Hussain, M.M.; Bakillah, A.; Jamil, H. Apolipoprotein B binding to microsomal triglyceride transfer protein decreases with increases in length and lipidation: Implications in lipoprotein biosynthesis. Biochemistry 1997, 36, 13060–13067. [Google Scholar] [CrossRef]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Conlon, D.M.; Thomas, T.; Fedotova, T.; Hernandez-Ono, A.; Di Paolo, G.; Chan, R.B.; Ruggles, K.; Gibeley, S.; Liu, J.; Ginsberg, H.N. Inhibition of apolipoprotein B synthesis stimulates endoplasmic reticulum autophagy that prevents steatosis. J. Clin. Investig. 2016, 126, 3852–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.-W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krahmer, N.; Farese, R.V., Jr.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Campbell–Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Atshaves, B.P.; McIntosh, A.M.; Lyuksyutova, O.I.; Zipfel, W.; Webb, W.W.; Schroeder, F. Liver fatty acid-binding protein gene ablation inhibits branched-chain fatty acid metabolism in cultured primary hepatocytes. J. Biol. Chem. 2004, 279, 30954–30965. [Google Scholar] [CrossRef] [Green Version]
- Hostetler, H.A.; Lupas, D.; Tan, Y.; Dai, J.; Kelzer, M.S.; Martin, G.G.; Woldegiorgis, G.; Kier, A.B.; Schroeder, F. Acyl-CoA binding proteins interact with the acyl-CoA binding domain of mitochondrial carnitine palmitoyl transferase I. Mol. Cell. Biochem. 2011, 355, 135–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusarova, V.; Brodsky, J.L.; Fisher, E.A. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J. Biol. Chem. 2003, 278, 48051–48058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Jacobs, R.L.; Devlin, C.; Tabas, I.; Vance, D.E. Targeted deletion of hepatic CTP: Phosphocholine cytidylyltransferase α in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 2004, 279, 47402–47410. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.L.; Lingrell, S.; Zhao, Y.; Francis, G.A.; Vance, D.E. Hepatic CTP: Phosphocholine cytidylyltransferase-α is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J. Biol. Chem. 2008, 283, 2147–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noga, A.A.; Zhao, Y.; Vance, D.E. An unexpected requirement for phosphatidylethanolaminen-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 2002, 277, 42358–42365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.-C.; Touboul, D.; Bertrand-Michel, J. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [Green Version]
- Grillet, M.; Gonzalez, B.D.; Sicart, A.; Pöttler, M.; Cascalho, A.; Billion, K.; Diaz, S.H.; Swerts, J.; Naismith, T.V.; Gounko, N.V. Torsins are essential regulators of cellular lipid metabolism. Dev. Cell 2016, 38, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Cascalho, A.; Foroozandeh, J.; Hennebel, L.; Swerts, J.; Klein, C.; Rous, S.; Dominguez Gonzalez, B.; Pisani, A.; Meringolo, M.; Gallego, S.F. Excess Lipin enzyme activity contributes to TOR1A recessive disease and DYT-TOR1A dystonia. Brain 2020, 143, 1746–1765. [Google Scholar] [CrossRef] [PubMed]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.-H.; Kwon, C.H.; Lee, K.-W.; Lee, J.-H.; Park, C.K. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res. 2008, 49, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Liu, Z.-X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.-m.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Muchir, A.; Wu, W.; Worman, H.J. Reduced expression of A-type lamins and emerin activates extracellular signal-regulated kinase in cultured cells. Biochim. Biophys. Acta 2009, 1792, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Maraldi, N.M.; Capanni, C.; Cenni, V.; Fini, M.; Lattanzi, G. Laminopathies and lamin-associated signaling pathways. J. Cell Biol. 2011, 112, 979–992. [Google Scholar] [CrossRef]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Luyendyk, J.P.; Tawfik, O.; Guo, G.L. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J. Pharmacol. Exp. Ther. 2009, 328, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S. Elafibranor, an agonist of the peroxisome proliferator—Activated receptor—α and—δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin. Investig. Drugs 2018, 27, 301–311. [Google Scholar] [CrossRef]
- Lefebvre, E.; Gottwald, M.; Lasseter, K.; Chang, W.; Willett, M.; Smith, P.; Somasunderam, A.; Utay, N. Pharmacokinetics, safety, and CCR2/CCR5 antagonist activity of cenicriviroc in participants with mild or moderate hepatic impairment. Clin. Transl. Sci. 2016, 9, 139–148. [Google Scholar] [CrossRef]
- Andrés, V.; González, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroen. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef]
- Shen, J.; Wang, S.; Zhang, Y.J.; Kappil, M.; Wu, H.C.; Kibriya, M.G.; Wang, Q.; Jasmine, F.; Ahsan, H.; Lee, P.H. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 2012, 55, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.-W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2017, 54, 78–88. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Kind, J.; van Steensel, B. Genome–nuclear lamina interactions and gene regulation. Curr. Opin. Cell Biol. 2010, 22, 320–325. [Google Scholar] [CrossRef]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 2017, 171, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Cesarini, E.; Mozzetta, C.; Marullo, F.; Gregoretti, F.; Gargiulo, A.; Columbaro, M.; Cortesi, A.; Antonelli, L.; Di Pelino, S.; Squarzoni, S. Lamin A/C sustains PcG protein architecture, maintaining transcriptional repression at target genes. J. Cell Biol. 2015, 211, 533–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.L.; Foisner, R. Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000554. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | Reported Year | No. of Affected Individuals | Mutation in TOR1AIP1 | Resultant LAP1 Protein | Phenotypes |
|---|---|---|---|---|---|
| Kayman-Kurekci et al. [70] | 2014 | 3 | c.186delG/c.186delG | p.E62fs*25 (truncation at 83 aa of LAP1B but intact LAP1C) | muscular dystrophy, joint contracture, cardiomyopathy |
| Dorboz et al. [71] | 2014 | 1 | c.1448A > T/c.1448A > T | p.E482A (E to A change in both LAP1 isoforms) | cerebellar atrophy, dystonia, cardiomyopathy, early death |
| Ghaoui et al. [72] | 2016 | 2 | c.127delC/c.1181T > C | p.P43fs*15/p.L394P (truncation at 58 aa of LAP1B, L to P change in both LAP1 isoforms) | muscular dystrophy, cardiomyopathy |
| Fichtman et al. [73] | 2019 | 7 | c.961C > T/c. 961C > T | p.R321* (truncation at 321 aa of both LAP1 isoforms) | multisytemic abnormalities, early death |
| Lessel et al. [74] | 2020 | 2 | c.945_948delCAGT/c.1331G > C | p.Q315fs*9/p.R444P (truncation at 315 aa and R to P changes in both LAP1 isoforms) | congenital hearing loss, developmental delay, brain abnormality |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Östlund, C.; Hernandez-Ono, A.; Shin, J.-Y. The Nuclear Envelope in Lipid Metabolism and Pathogenesis of NAFLD. Biology 2020, 9, 338. https://doi.org/10.3390/biology9100338
Östlund C, Hernandez-Ono A, Shin J-Y. The Nuclear Envelope in Lipid Metabolism and Pathogenesis of NAFLD. Biology. 2020; 9(10):338. https://doi.org/10.3390/biology9100338
Chicago/Turabian StyleÖstlund, Cecilia, Antonio Hernandez-Ono, and Ji-Yeon Shin. 2020. "The Nuclear Envelope in Lipid Metabolism and Pathogenesis of NAFLD" Biology 9, no. 10: 338. https://doi.org/10.3390/biology9100338
APA StyleÖstlund, C., Hernandez-Ono, A., & Shin, J.-Y. (2020). The Nuclear Envelope in Lipid Metabolism and Pathogenesis of NAFLD. Biology, 9(10), 338. https://doi.org/10.3390/biology9100338