Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets for Nuclear, Cytoplasmic and Nucleocytoplasmic Proteins
2.2. NLS and NES Prediction
2.3. Identification of Novel Motifs with MEME and SLiMSearch
2.4. Proteomics Analysis
3. Results
3.1. Many Nuclear Localized Proteins Do Not Have a Predictable cNLS
3.2. Many Imp-α Binding Partners Do Not Have a Predictable cNLS
3.3. Identification of Putative Piggybacking Proteins
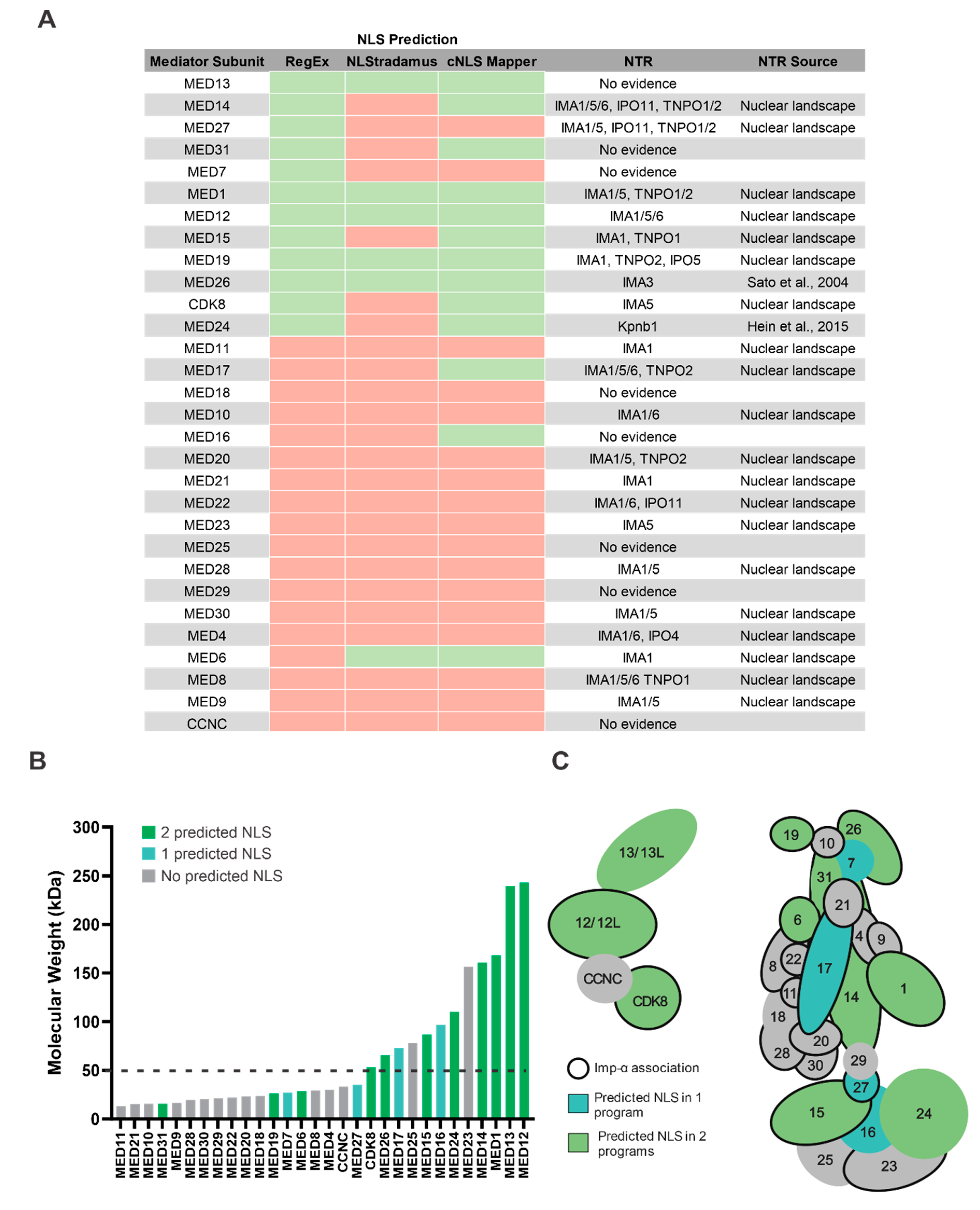
3.4. Mediator Proteins Associate with Imp-α and Do Not Have a Predictable cNLS
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Hoelz, A.; Debler, E.W.; Blobel, G. The Structure of the Nuclear Pore Complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knockenhauer, K.E.; Schwartz, T.U. The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell 2016, 164, 1162–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.B.; Görlich, D. Transport Selectivity of Nuclear Pores, Phase Separation, and Membraneless Organelles. Trends Biochem. Sci. 2016, 41, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Frey, S.; Fischer, T.; Güttler, T.; Görlich, D. Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009, 28, 2541–2553. [Google Scholar] [CrossRef] [Green Version]
- Frey, S.; Rees, R.; Schunemann, J.; Ng, S.C.; Funfgeld, K.; Huyton, T.; Gorlich, D. Surface Properties Determining Passage Rates of Proteins through Nuclear Pores. Cell 2018, 174, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Ribbeck, K.; Lipowsky, G.; Kent, H.M.; Stewart, M.; Görlich, D. NTF2 mediates nuclear import of Ran. EMBO J. 1998, 17, 6587–6598. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.F.; Li, S.; Chen, Y.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J. Biol. Chem. 1996, 271, 32863–32868. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, A.J.; Dacks, J.B.; Field, M.C. Evolution of the karyopherin-β family of nucleocytoplasmic transport factors; ancient origins and continued specialization. PLoS ONE 2011, 6, e19308. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M.; Imamoto, N. Biological significance of the importin-β family-dependent nucleocytoplasmic transport pathways. Traffic 2014, 15, 727–748. [Google Scholar] [CrossRef]
- Soniat, M.; Chook, Y.M. Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem. J. 2015, 468, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Meinkoth, J.L.; Tsien, R.Y.; Taylor, S.S. Identification of a signal for rapid export of proteins from the nucleus. Cell 1995, 82, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Cautain, B.; Hill, R.; de Pedro, N.; Link, W. Components and regulation of nuclear transport processes. FEBS J. 2015, 282, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Görlich, D.; Kostka, S.; Kraft, R.; Dingwall, C.; Laskey, R.A.; Hartmann, E.; Prehn, S. Two different subunits of importin cooperate to recognize nuclear localization signals and bind them to the nuclear envelope. Curr. Biol. 1995, 5, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, D.S.; Corbett, A.H.; Mason, D.A.; Harreman, M.T.; Adam, S.A. Importin alpha: A multipurpose nuclear-transport receptor. Trends Cell Biol. 2004, 14, 505–514. [Google Scholar] [CrossRef]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical Nuclear Localization Signals: Definition, Function, and Interaction with Importin. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [Green Version]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Robbins, J.; Dilworth, S.M.; Laskey, R.A.; Dingwall, C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: Identification of a class of bipartite nuclear targeting sequence. Cell 1991, 64, 615–623. [Google Scholar] [CrossRef]
- Conti, E.; Uy, M.; Leighton, L.; Blobel, G.; Kuriyan, J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell 1998, 94, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Conti, E.; Kuriyan, J. Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure 2000, 8, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Lott, K.; Bhardwaj, A.; Sims, P.J.; Cingolani, G. A minimal nuclear localization signal (NLS) in human phospholipid scramblase 4 that binds only the minor NLS-binding site of importin alpha1. J. Biol. Chem. 2011, 286, 28160–28169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; Zhou, H.-X. Design rules for selective binding of nuclear localization signals to minor site of importin alpha. PLoS ONE 2014, 9, e91025. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Hasebe, M.; Matsumura, N.; Takashima, H.; Miyamoto-Sato, E.; Tomita, M.; Yanagawa, H. Six classes of nuclear localization signals specific to different binding grooves of importin α. J. Biol. Chem. 2009, 284, 478–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, R.; Hirano, H.; Matsuura, Y. Structure of importin-alpha bound to a non-classical nuclear localization signal of the influenza A virus nucleoprotein. Sci. Rep. 2015, 5, 15055. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Cansizoglu, A.E.; Suel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, G.N.; Cook, N.J.; Wang, W.; Hare, S.; Gupta, S.S.; Öztop, I.; Lee, K.; Pye, V.E.; Cosnefroy, O.; Snijders, A.P.; et al. Structural basis for nuclear import of splicing factors by human Transportin 3. Proc. Natl. Acad. Sci. USA 2014, 111, 2728–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornerod, M.; Ohno, M.; Yoshida, M.; Mattaj, I.W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 1997, 90, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311. [Google Scholar] [CrossRef]
- Dong, X.; Biswas, A.; Süel, K.E.; Jackson, L.K.; Martinez, R.; Gu, H.; Chook, Y.M. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 2009, 458, 1136–1141. [Google Scholar] [CrossRef]
- Kirli, K.; Karaca, S.; Dehne, H.J.; Samwer, M.; Pan, K.T.; Lenz, C.; Urlaub, H.; Görlich, D. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. eLife 2015, 4, e11466. [Google Scholar] [CrossRef]
- Bernhofer, M.; Goldberg, T.; Wolf, S.; Ahmed, M.; Zaugg, J.; Boden, M.; Rost, B. NLSdb—Major update for database of nuclear localization signals and nuclear export signals. Nucleic Acids Res. 2017, 46, D503–D508. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Ba, A.N.; Pogoutse, A.; Provart, N.; Moses, A.M. NLStradamus: A simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinform. 2009, 10, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brameier, M.; Krings, A.; MacCallum, R.M. NucPred—Predicting nuclear localization of proteins. Bioinformatics 2007, 23, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Hu, J. SeqNLS: Nuclear Localization Signal Prediction Based on Frequent Pattern Mining and Linear Motif Scoring. PLoS ONE 2013, 8, e76864. [Google Scholar] [CrossRef]
- Marfori, M.; Mynott, A.; Ellis, J.J.; Mehdi, A.M.; Saunders, N.F.W.; Curmi, P.M.; Forwood, J.K.; Bodén, M.; Kobe, B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 2011, 1813, 1562–1577. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, 3321. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.-K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2016, 45, D1100–D1106. [Google Scholar] [CrossRef]
- Trowitzsch, S.; Viola, C.; Scheer, E.; Conic, S.; Chavant, V.; Fournier, M.; Papai, G.; Ebong, I.-O.; Schaffitzel, C.; Zou, J.; et al. Cytoplasmic TAF2-TAF8-TAF10 complex provides evidence for nuclear holo-TFIID assembly from preformed submodules. Nat. Commun. 2015, 6, 6011. [Google Scholar] [CrossRef] [Green Version]
- Asally, M.; Yoneda, Y. Beta-catenin can act as a nuclear import receptor for its partner transcription factor, lymphocyte enhancer factor-1 (lef-1). Exp. Cell Res. 2005, 308, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Czeko, E.; Seizl, M.; Augsberger, C.; Mielke, T.; Cramer, P. Iwr1 Directs RNA Polymerase II Nuclear Import. Mol. Cell 2011, 42, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Croce, L. Regulating the Shuttling of Eukaryotic RNA Polymerase II. Mol. Cell. Biol. 2011, 31, 3918–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bange, G.; Murat, G.; Sinning, I.; Hurt, E.; Kressler, D. New twist to nuclear import: When two travel together. Commun. Integr. Biol. 2013, 6, e24792. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Bange, G.; Ogawa, Y.; Stjepanovic, G.; Bradatsch, B.; Pratte, D.; Amlacher, S.; Strauß, D.; Yoneda, Y.; Katahira, J.; et al. Synchronizing nuclear import of ribosomal proteins with ribosome assembly. Science 2012, 338, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Čalyševa, J.; et al. ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Krystkowiak, I.; Davey, N.E. SLiMSearch: A framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Mackmull, M.; Klaus, B.; Heinze, I.; Chokkalingam, M.; Beyer, A.; Russell, R.B.; Ori, A.; Beck, M. Landscape of nuclear transport receptor cargo specificity. Mol. Syst. Biol. 2017, 13, 962. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Teo, G.; Liu, G.; Zhang, J.; Nesvizhskii, A.I.; Gingras, A.-C.; Choi, H. SAINTexpress: Improvements and additional features in Significance Analysis of INTeractome software. J. Proteom. 2014, 100, 37–43. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.D.R.; Choi, H.; Gupta, G.D.; Pelletier, L.; Raught, B.; Nesvizhskii, A.I.; Gingras, A.-C. ProHits-viz: A suite of web tools for visualizing interaction proteomics data. Nat. Methods 2017, 14, 645–646. [Google Scholar] [CrossRef]
- Xu, D.; Farmer, A.; Chook, Y.M. Recognition of nuclear targeting signals by Karyopherin-beta proteins. Curr. Opin. Struct. Biol. 2010, 20, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.-Q.; Dasari, S.; Chambers, M.C.; Litton, M.D.; Sobecki, S.M.; Zimmerman, L.J.; Halvey, P.J.; Schilling, B.; Drake, P.M.; Gibson, B.W.; et al. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteom. Res. 2009, 8, 3872–3881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.C.; Shimizu, Y.; Minoshima, S.; Shimizu, N.; Haussler, C.A.; Jurutka, P.W.; Haussler, M.R. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J. Cell. Biochem. 1998, 70, 94–109. [Google Scholar] [CrossRef]
- Prufer, K.; Barsony, J. Retinoid X Receptor Dominates the Nuclear Import and Export of the Unliganded Vitamin D Receptor. Mol. Endocrinol. 2002, 16, 1738–1751. [Google Scholar] [CrossRef]
- Chopin-Delannoy, S.; Thénot, S.; Delaunay, F.; Buisine, E.; Begue, A.; Duterque-Coquillaud, M.; Laudet, V. A specific and unusual nuclear localization signal in the DNA binding domain of the Rev-erb orphan receptors. J. Mol. Endocrinol. 2003, 30, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Boulon, S.; Pradet-Balade, B.; Verheggen, C.; Molle, D.; Boireau, S.; Georgieva, M.; Azzag, K.; Robert, M.-C.; Ahmad, Y.; Neel, H.; et al. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell 2010, 39, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Marko, M.; Vlassis, A.; Guialis, A.; Leichter, M. Domains involved in TAF15 subcellular localisation: Dependence on cell type and ongoing transcription. Gene 2012, 506, 331–338. [Google Scholar] [CrossRef]
- Soniat, M.; Chook, Y.M. Karyopherin-beta2 Recognition of a PY-NLS Variant that Lacks the Proline-Tyrosine Motif. Structure 2016, 24, 1802–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soniat, M.; Sampathkumar, P.; Collett, G.; Gizzi, A.S.; Banu, R.N.; Bhosle, R.C.; Chamala, S.; Chowdhury, S.; Fiser, A.; Glenn, A.S.; et al. Crystal structure of human Karyopherin β2 bound to the PY-NLS of Saccharomyces cerevisiae Nab2. J. Struct. Funct. Genom. 2013, 14, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Gama-Carvalho, M.; Carmo-Fonseca, M. The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett. 2001, 498, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E.; Haslam, N.J.; Shields, D.C.; Edwards, R.J. SLiMFinder: A web server to find novel, significantly over-represented, short protein motifs. Nucleic Acids Res. 2010, 38, W534–W539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neduva, V.; Linding, R.; Su-Angrand, I.; Stark, A.; De Masi, F.; Gibson, T.J.; Lewis, J.; Serrano, L.; Russell, R.B. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Gibson, T.J.; Dinkel, H.; Van Roey, K.; Diella, F. Experimental detection of short regulatory motifs in eukaryotic proteins: Tips for good practice as well as for bad. Cell Commun. Signal. 2015, 13, 42. [Google Scholar] [CrossRef] [Green Version]
- Arjomand, A.; Baker, M.A.; Li, C.; Buckle, A.M.; Jans, D.A.; Loveland, K.L.; Miyamoto, Y. The α-importome of mammalian germ cell maturation provides novel insights for importin biology. FASEB J. 2014, 28, 3480–3493. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019, 48, 44–54. [Google Scholar] [CrossRef]
- Kamenova, I.; Mukherjee, P.; Conic, S.; Mueller, F.; El-Saafin, F.; Bardot, P.; Garnier, J.-M.; Dembele, D.; Capponi, S.; Timmers, H.T.M.; et al. Co-translational assembly of mammalian nuclear multisubunit complexes. Nat. Commun. 2019, 10, 1740. [Google Scholar] [CrossRef] [Green Version]
- Soutoglou, E.; Demény, M.A.; Scheer, E.; Fienga, G.; Sassone-Corsi, P.; Tora, L. The nuclear import of TAF10 is regulated by one of its three histone fold domain-containing interaction partners. Mol. Cell. Biol. 2005, 25, 4092–4104. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; Greber, B.J.; Nogales, E. Recent insights into the structure of TFIID, its assembly, and its binding to core promoter. Curr. Opin. Struct. Biol. 2020, 61, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Antonova, S.V.; Haffke, M.; Corradini, E.; Mikuciunas, M.; Low, T.Y.; Signor, L.; van Es, R.M.; Gupta, K.; Scheer, E.; Vos, H.R.; et al. Chaperonin CCT checkpoint function in basal transcription factor TFIID assembly. Nat. Struct. Mol. Biol. 2018, 25, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, H.-M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008, 36, 3993–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Poss, Z.C.; Ebmeier, C.C.; Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575–608. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.-L.; Tomomori-Sato, C.; Sato, S.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. Subunit architecture and functional modular rearrangements of the transcriptional mediator complex. Cell 2014, 157, 1430–1444. [Google Scholar] [CrossRef] [Green Version]
- Lolodi, O.; Yamazaki, H.; Otsuka, S.; Kumeta, M.; Yoshimura, S.H. Dissecting in vivo steady-state dynamics of karyopherin-dependent nuclear transport. Mol. Biol. Cell 2016, 27, 167–176. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessier, T.M.; MacNeil, K.M.; Mymryk, J.S. Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome. Biology 2020, 9, 188. https://doi.org/10.3390/biology9080188
Tessier TM, MacNeil KM, Mymryk JS. Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome. Biology. 2020; 9(8):188. https://doi.org/10.3390/biology9080188
Chicago/Turabian StyleTessier, Tanner M., Katelyn M. MacNeil, and Joe S. Mymryk. 2020. "Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome" Biology 9, no. 8: 188. https://doi.org/10.3390/biology9080188
APA StyleTessier, T. M., MacNeil, K. M., & Mymryk, J. S. (2020). Piggybacking on Classical Import and Other Non-Classical Mechanisms of Nuclear Import Appear Highly Prevalent within the Human Proteome. Biology, 9(8), 188. https://doi.org/10.3390/biology9080188