

Evaluation of Teneligliptin and Retagliptin on the Clearance of Melanosome by Melanophagy in B16F1 Cells

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Cell Based Library Screening
2.4. Assay for Melanin Content
2.5. Autophagy Analysis
2.6. Melanophagy Assay
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
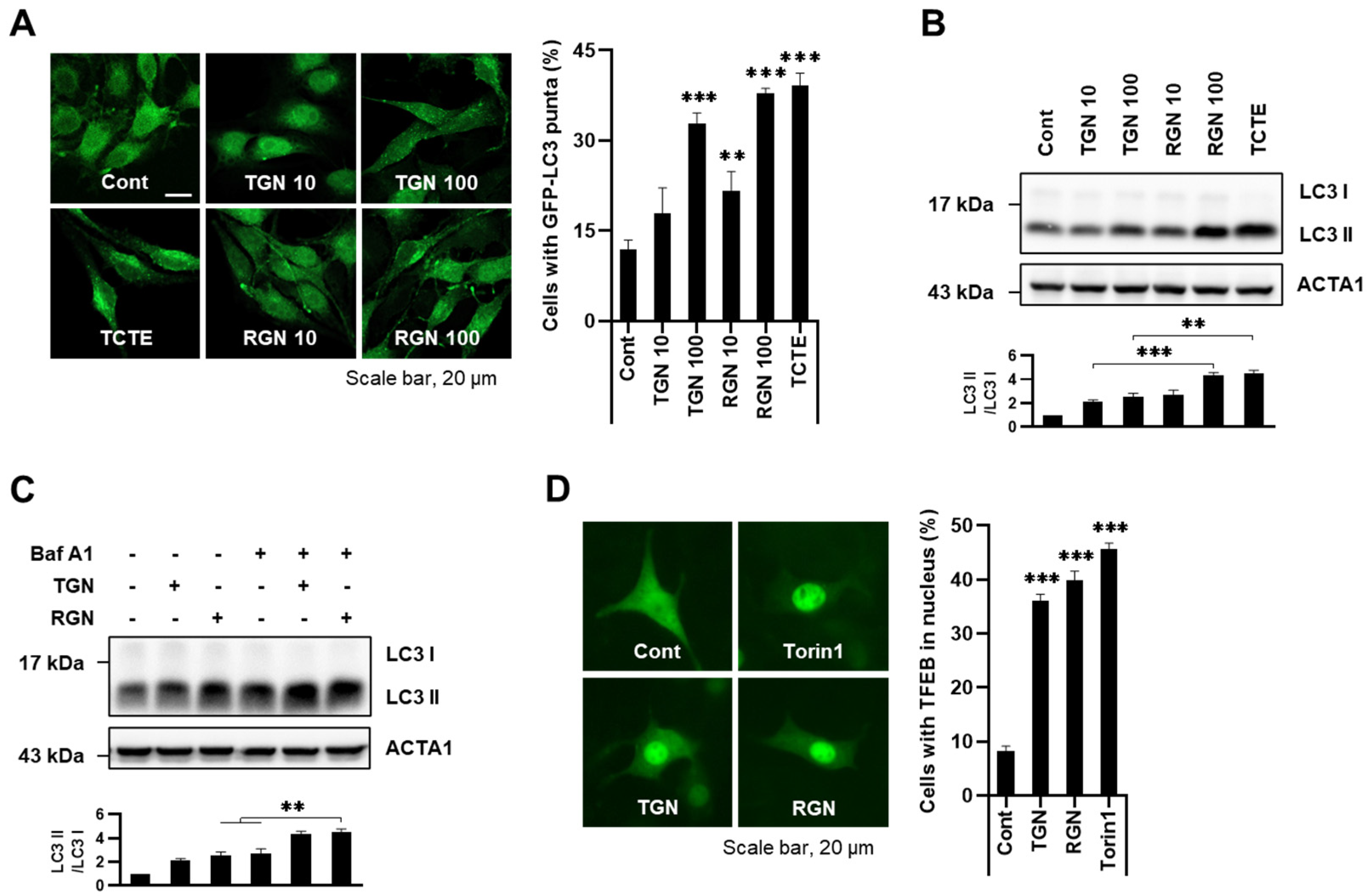
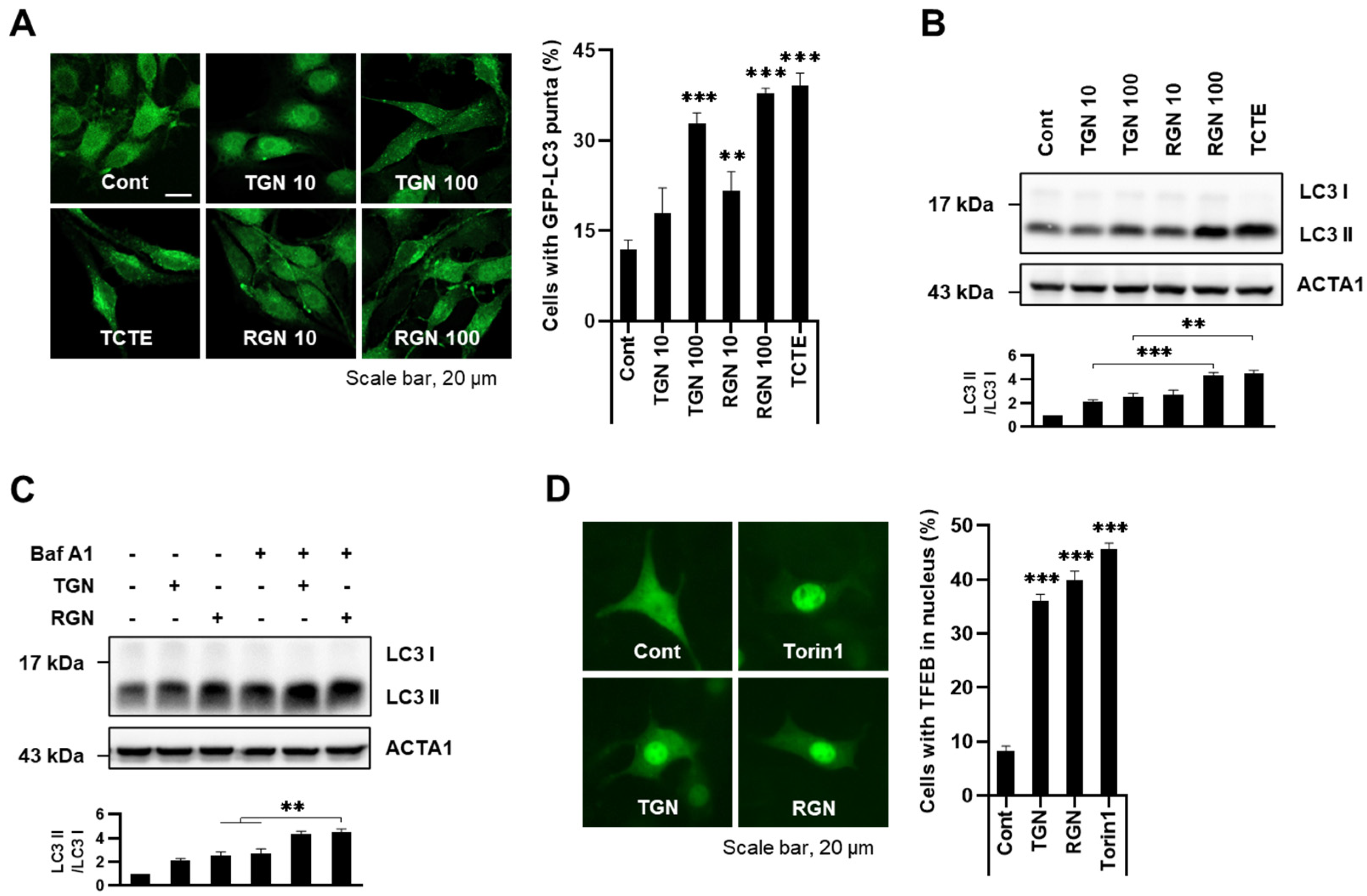
3.1. Teneligliptin Hydrobromide and Retagliptin Phosphate Activate Autophagy in B16F1 Cells
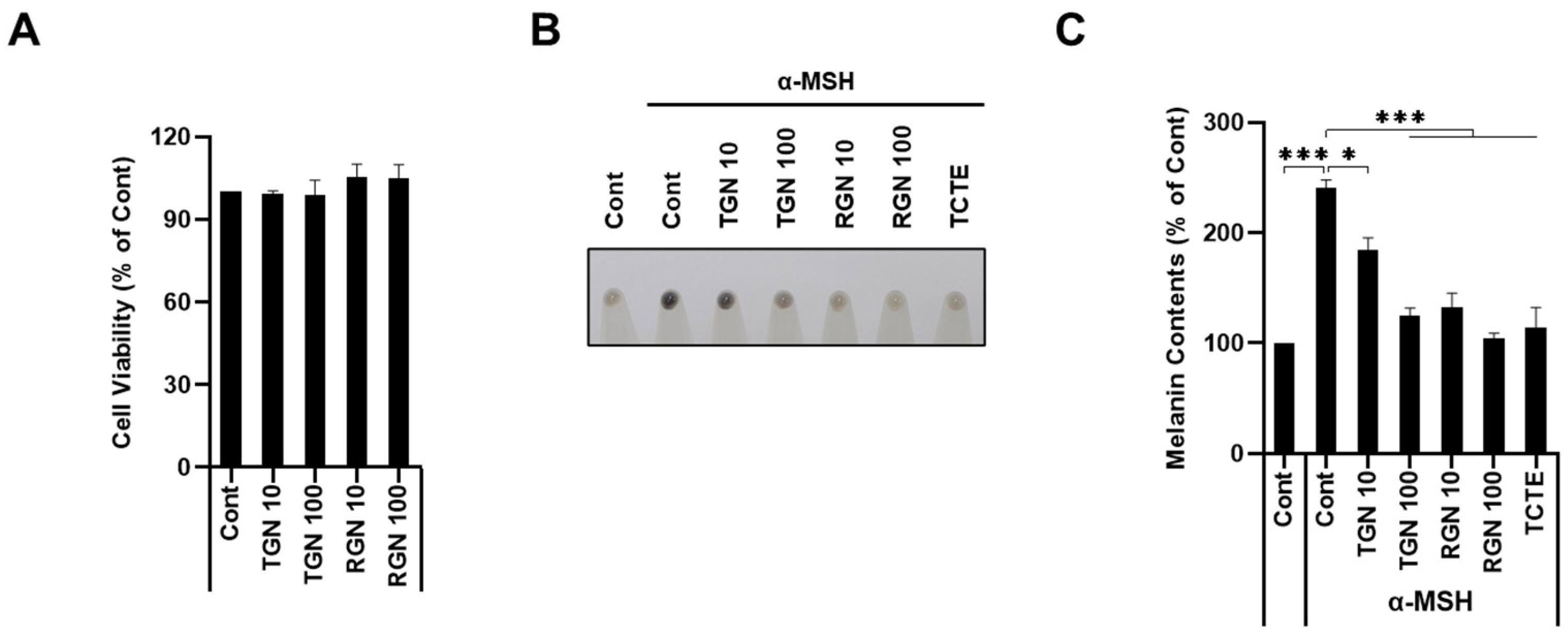
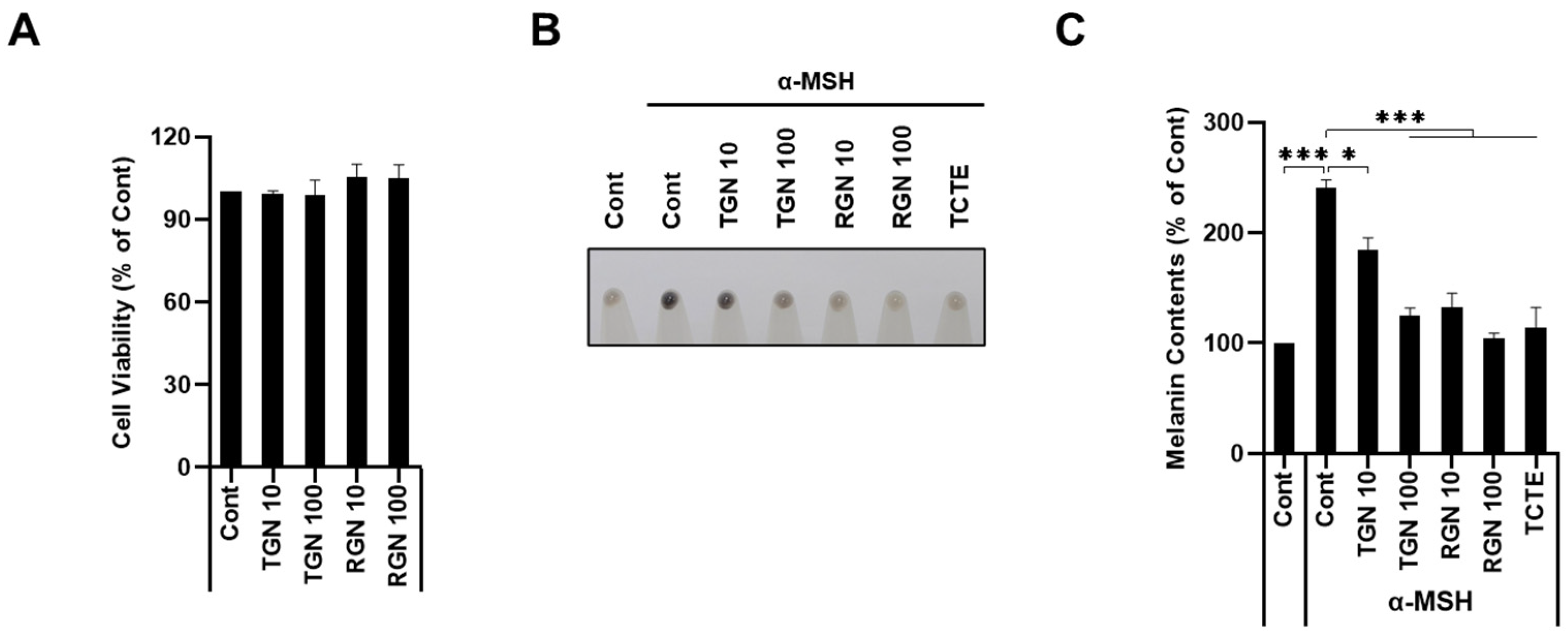
3.2. Teneligliptin Hydrobromide and Retagliptin Phosphate Inhibit α-MSH-Stimulated Melanogenesis in B16F1 Cells
3.3. Teneligliptin Hydrobromide and Retagliptin Phosphate Promote Melanophagy in B16F1 Cells
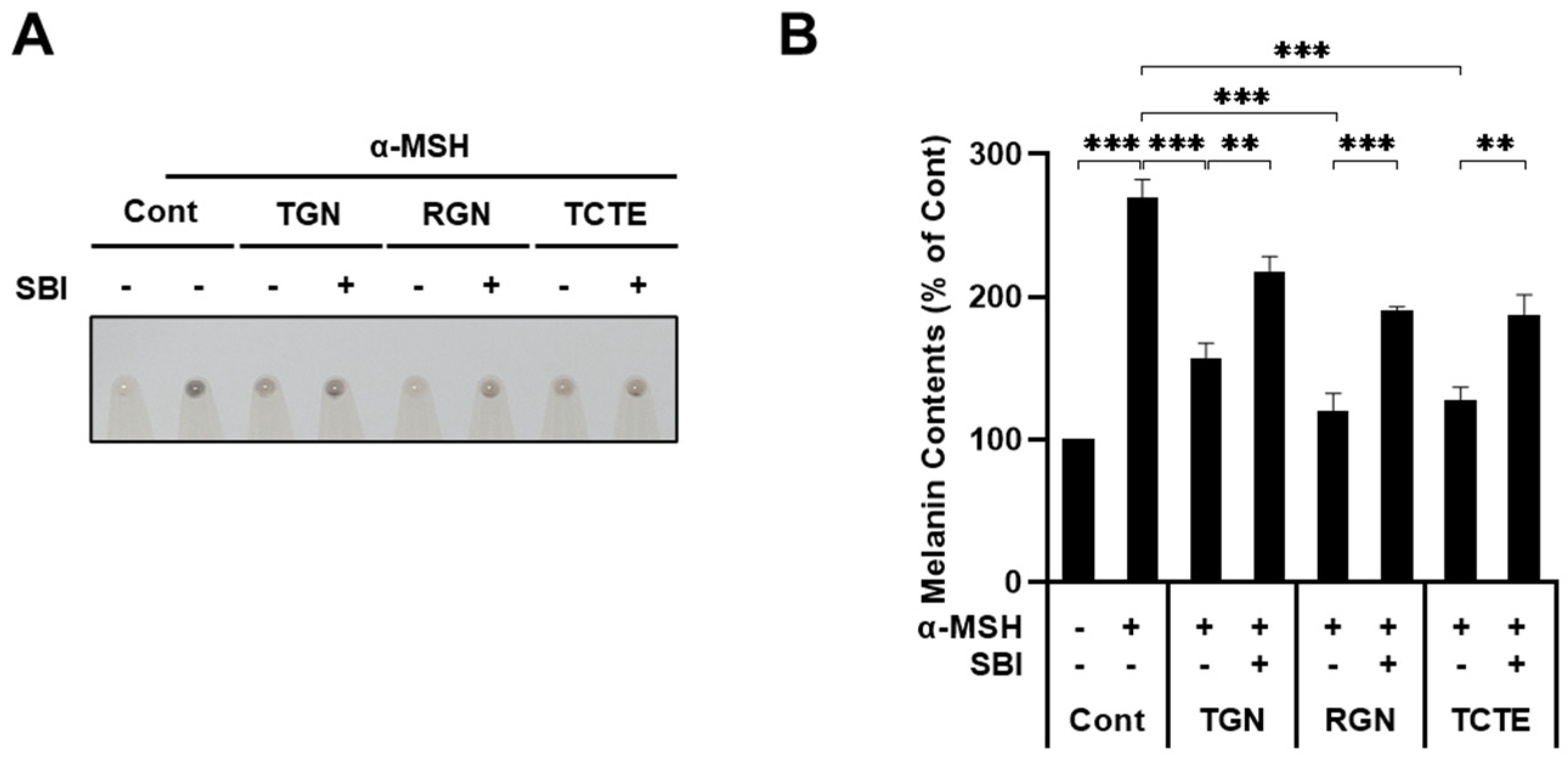
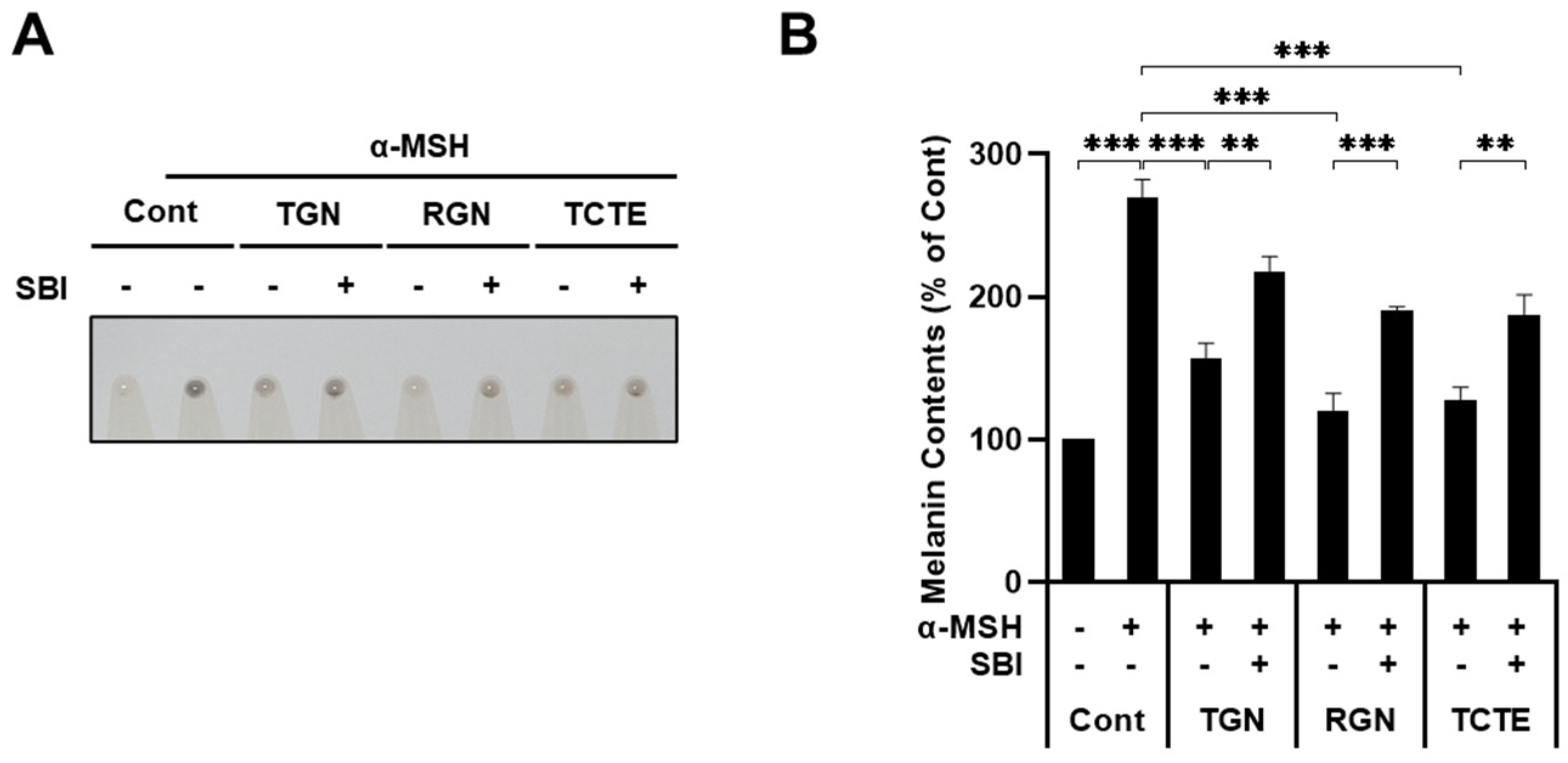
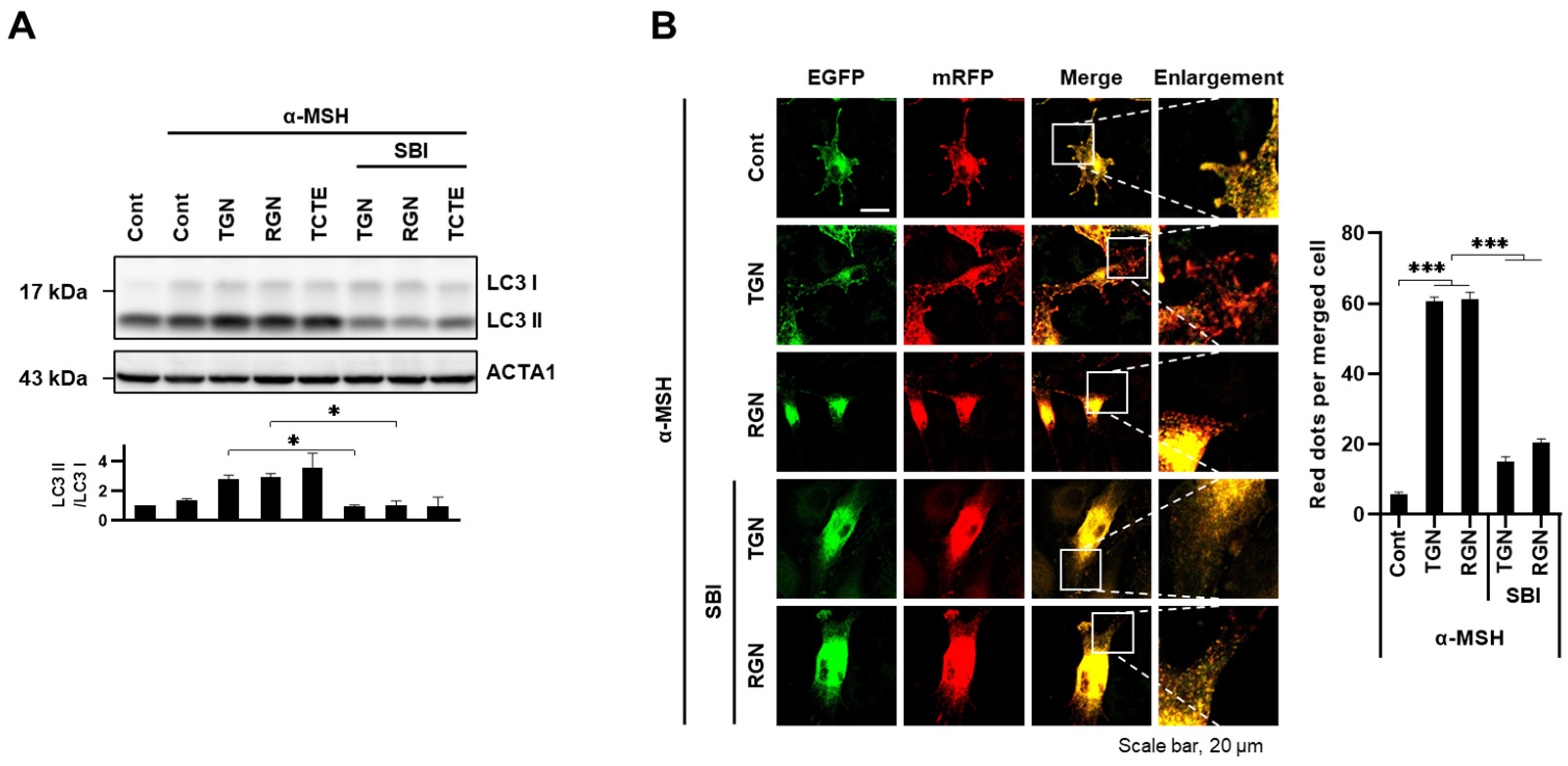
3.4. ULK1 Inhibitor Decreases Melanophagy Induced by Teneligliptin Hydrobromide and Retagliptin Phosphate
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Johansen, T.; Dikic, I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [PubMed]
- von Muhlinen, N.; Thurston, T.; Ryzhakov, G.; Bloor, S.; Randow, F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy 2010, 6, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Shah, H.V.; Kanfer, G.; Pickrell, A.M.; Holtzclaw, L.A.; Ward, M.E.; Youle, R.J. Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol. Cell 2020, 80, 779–795.e710. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ji, L.; Hu, J.; Zhao, Y.; Johnston, L.J.; Zhang, X.; Ma, X. Functional Amino Acids and Autophagy: Diverse Signal Transduction and Application. Int. J. Mol. Sci. 2021, 22, 11427. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef]
- Hofmann, E.; Schwarz, A.; Fink, J.; Kamolz, L.P.; Kotzbeck, P. Modelling the Complexity of Human Skin In Vitro. Biomedicines 2023, 11, 794. [Google Scholar] [CrossRef]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef]
- Le, L.; Sires-Campos, J.; Raposo, G.; Delevoye, C.; Marks, M.S. Melanosome Biogenesis in the Pigmentation of Mammalian Skin. Integr. Comp. Biol. 2021, 61, 1517–1545. [Google Scholar] [CrossRef]
- Bissig, C.; Rochin, L.; van Niel, G. PMEL Amyloid Fibril Formation: The Bright Steps of Pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Kamiya, T.; Kawakami, A.; Ogino, J.; Sohma, H.; Uhara, H.; Jimbow, K. Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma. Int. J. Mol. Sci. 2020, 21, 6129. [Google Scholar] [CrossRef]
- Kovacs, D.; Cardinali, G.; Picardo, M.; Bastonini, E. Shining Light on Autophagy in Skin Pigmentation and Pigmentary Disorders. Cells 2022, 11, 2999. [Google Scholar] [CrossRef]
- Zhang, C.F.; Gruber, F.; Ni, C.; Mildner, M.; Koenig, U.; Karner, S.; Barresi, C.; Rossiter, H.; Narzt, M.S.; Nagelreiter, I.M.; et al. Suppression of autophagy dysregulates the antioxidant response and causes premature senescence of melanocytes. J. Investig. Dermatol. 2015, 135, 1348–1357. [Google Scholar] [CrossRef]
- Park, H.J.; Jo, D.S.; Choi, D.S.; Bae, J.E.; Park, N.Y.; Kim, J.B.; Chang, J.H.; Shin, J.J.; Cho, D.H. Ursolic acid inhibits pigmentation by increasing melanosomal autophagy in B16F1 cells. Biochem. Biophys. Res. Commun. 2020, 531, 209–214. [Google Scholar] [CrossRef]
- Park, H.J.; Jo, D.S.; Choi, H.; Bae, J.E.; Park, N.Y.; Kim, J.B.; Choi, J.Y.; Kim, Y.H.; Oh, G.S.; Chang, J.H.; et al. Melasolv induces melanosome autophagy to inhibit pigmentation in B16F1 cells. PLoS ONE 2020, 15, e0239019. [Google Scholar] [CrossRef]
- Tsao, Y.T.; Huang, Y.F.; Kuo, C.Y.; Lin, Y.C.; Chiang, W.C.; Wang, W.K.; Hsu, C.W.; Lee, C.H. Hinokitiol Inhibits Melanogenesis via AKT/mTOR Signaling in B16F10 Mouse Melanoma Cells. Int. J. Mol. Sci. 2016, 17, 248. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Jo, Y.K.; Park, S.J.; Chang, H.; Shin, J.H.; Choi, E.S.; Kim, J.B.; Seok, S.H.; Kim, J.S.; Oh, J.S.; et al. ARP101 inhibits alpha-MSH-stimulated melanogenesis by regulation of autophagy in melanocytes. FEBS Lett. 2013, 587, 3955–3960. [Google Scholar] [CrossRef] [PubMed]
- Zou, N.; Wei, Y.; Li, F.; Yang, Y.; Cheng, X.; Wang, C. The inhibitory effects of compound Muniziqi granule against B16 cells and harmine induced autophagy and apoptosis by inhibiting Akt/mTOR pathway. BMC Complement. Altern. Med. 2017, 17, 517. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, M.; Chen, L.; Zhou, L.; Bian, S.; Lv, Y. Licochalcone A restrains microphthalmia-associated transcription factor expression and growth by activating autophagy in melanoma cells via miR-142-3p/Rheb/mTOR pathway. Phytother. Res. 2020, 34, 349–358. [Google Scholar] [CrossRef]
- Yoon, J.H.; Youn, K.; Jun, M. Discovery of Pinostrobin as a Melanogenic Agent in cAMP/PKA and p38 MAPK Signaling Pathway. Nutrients 2022, 14, 3713. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, S.H.; Kim, Y.H.; Kim, S.H.; Oh, G.S.; Bae, J.E.; Kim, J.B.; Park, N.Y.; Park, K.; Yeom, E.; et al. Nalfurafine Hydrochloride, a kappa-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells. Cells 2022, 12, 146. [Google Scholar] [CrossRef]
- Bento-Lopes, L.; Cabaco, L.C.; Charneca, J.; Neto, M.V.; Seabra, M.C.; Barral, D.C. Melanin’s Journey from Melanocytes to Keratinocytes: Uncovering the Molecular Mechanisms of Melanin Transfer and Processing. Int. J. Mol. Sci. 2023, 24, 11289. [Google Scholar] [CrossRef]
- Kang, H.H.; Rho, H.S.; Hwang, J.S.; Oh, S.G. Depigmenting activity and low cytotoxicity of alkoxy benzoates or alkoxy cinnamte in cultured melanocytes. Chem. Pharm. Bull. 2003, 51, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, T.; Kondoh, H.; Hiratsuka, J.; Mishima, Y. Enhanced melanogenesis induced by tyrosinase gene-transfer increases boron-uptake and killing effect of boron neutron capture therapy for amelanotic melanoma. Pigment. Cell Res. 1998, 11, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.W.; Schwartz, R.A.; Janniger, C.K. Melasma. G. Ital. Dermatol. Venereol. 2017, 152, 36–45. [Google Scholar] [CrossRef]
- Bellei, B.; Pitisci, A.; Ottaviani, M.; Ludovici, M.; Cota, C.; Luzi, F.; Dell’Anna, M.L.; Picardo, M. Vitiligo: A possible model of degenerative diseases. PLoS ONE 2013, 8, e59782. [Google Scholar] [CrossRef]
- Gutman, I.; Dunn, D.; Behrens, M.; Gold, A.P.; Odel, J.; Olarte, M.R. Hypopigmented iris spot. An early sign of tuberous sclerosis. Ophthalmology 1982, 89, 1155–1159. [Google Scholar] [CrossRef]
- Kim, Y.H.; Jo, D.S.; Park, N.Y.; Bae, J.E.; Kim, J.B.; Lee, H.J.; Kim, S.H.; Kim, S.H.; Lee, S.; Son, M.; et al. Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation. Cells 2022, 11, 2839. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Bluher, M.; Czech, M.P.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018, 555, 673–677. [Google Scholar] [CrossRef]
- Kong, L.; Deng, J.; Zhou, X.; Cai, B.; Zhang, B.; Chen, X.; Chen, Z.; Wang, W. Sitagliptin activates the p62-Keap1-Nrf2 signalling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. 2021, 12, 928. [Google Scholar] [CrossRef]
- Song, Y.; Yang, H.; Kim, J.; Lee, Y.; Kim, S.H.; Do, I.G.; Park, C.Y. Gemigliptin, a DPP4 inhibitor, ameliorates nonalcoholic steatohepatitis through AMP-activated protein kinase-independent and ULK1-mediated autophagy. Mol. Metab. 2023, 78, 101806. [Google Scholar] [CrossRef]
- Arab, H.H.; Gad, A.M.; Reda, E.; Yahia, R.; Eid, A.H. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: Targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021, 269, 119031. [Google Scholar] [CrossRef]
- Wesley, U.V.; Albino, A.P.; Tiwari, S.; Houghton, A.N. A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J. Exp. Med. 1999, 190, 311–322. [Google Scholar] [CrossRef]
- Pethiyagoda, C.L.; Welch, D.R.; Fleming, T.P. Dipeptidyl peptidase IV (DPPIV) inhibits cellular invasion of melanoma cells. Clin. Exp. Metastasis 2000, 18, 391–400. [Google Scholar] [CrossRef]
- Beckers, P.A.J.; Gielis, J.F.; Van Schil, P.E.; Adriaensen, D. Lung ischemia reperfusion injury: The therapeutic role of dipeptidyl peptidase 4 inhibition. Ann. Transl. Med. 2017, 5, 129. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Englaro, W.; Bahadoran, P.; Bertolotto, C.; Busca, R.; Derijard, B.; Livolsi, A.; Peyron, J.F.; Ortonne, J.P.; Ballotti, R. Tumor necrosis factor alpha-mediated inhibition of melanogenesis is dependent on nuclear factor kappa B activation. Oncogene 1999, 18, 1553–1559. [Google Scholar] [CrossRef]
- Fu, C.; Chen, J.; Lu, J.; Yi, L.; Tong, X.; Kang, L.; Pei, S.; Ouyang, Y.; Jiang, L.; Ding, Y.; et al. Roles of inflammation factors in melanogenesis (Review). Mol. Med. Rep. 2020, 21, 1421–1430. [Google Scholar] [CrossRef]
- Wang, X.; Ke, J.; Zhu, Y.J.; Cao, B.; Yin, R.L.; Wang, Y.; Wei, L.L.; Zhang, L.J.; Yang, L.Y.; Zhao, D. Dipeptidyl peptidase-4 (DPP4) inhibitor sitagliptin alleviates liver inflammation of diabetic mice by acting as a ROS scavenger and inhibiting the NFkappaB pathway. Cell Death Discov. 2021, 7, 236. [Google Scholar] [CrossRef]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The Role of Autophagy in Skin Fibroblasts, Keratinocytes, Melanocytes, and Epidermal Stem Cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef]
- Li, Y.F.; Ouyang, S.H.; Tu, L.F.; Wang, X.; Yuan, W.L.; Wang, G.E.; Wu, Y.P.; Duan, W.J.; Yu, H.M.; Fang, Z.Z.; et al. Caffeine Protects Skin from Oxidative Stress-Induced Senescence through the Activation of Autophagy. Theranostics 2018, 8, 5713–5730. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Bae, J.-E.; Park, N.Y.; Kim, J.B.; Kim, Y.H.; Kim, S.H.; Oh, G.S.; Wang, H.W.; Chang, J.H.; Cho, D.-H. Evaluation of Teneligliptin and Retagliptin on the Clearance of Melanosome by Melanophagy in B16F1 Cells. Cosmetics 2024, 11, 35. https://doi.org/10.3390/cosmetics11020035
Kim SH, Bae J-E, Park NY, Kim JB, Kim YH, Kim SH, Oh GS, Wang HW, Chang JH, Cho D-H. Evaluation of Teneligliptin and Retagliptin on the Clearance of Melanosome by Melanophagy in B16F1 Cells. Cosmetics. 2024; 11(2):35. https://doi.org/10.3390/cosmetics11020035
Chicago/Turabian StyleKim, Seong Hyun, Ji-Eun Bae, Na Yeon Park, Joon Bum Kim, Yong Hwan Kim, So Hyun Kim, Gyeong Seok Oh, Hee Won Wang, Jeong Ho Chang, and Dong-Hyung Cho. 2024. "Evaluation of Teneligliptin and Retagliptin on the Clearance of Melanosome by Melanophagy in B16F1 Cells" Cosmetics 11, no. 2: 35. https://doi.org/10.3390/cosmetics11020035
APA StyleKim, S. H., Bae, J.-E., Park, N. Y., Kim, J. B., Kim, Y. H., Kim, S. H., Oh, G. S., Wang, H. W., Chang, J. H., & Cho, D.-H. (2024). Evaluation of Teneligliptin and Retagliptin on the Clearance of Melanosome by Melanophagy in B16F1 Cells. Cosmetics, 11(2), 35. https://doi.org/10.3390/cosmetics11020035