Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories

Abstract
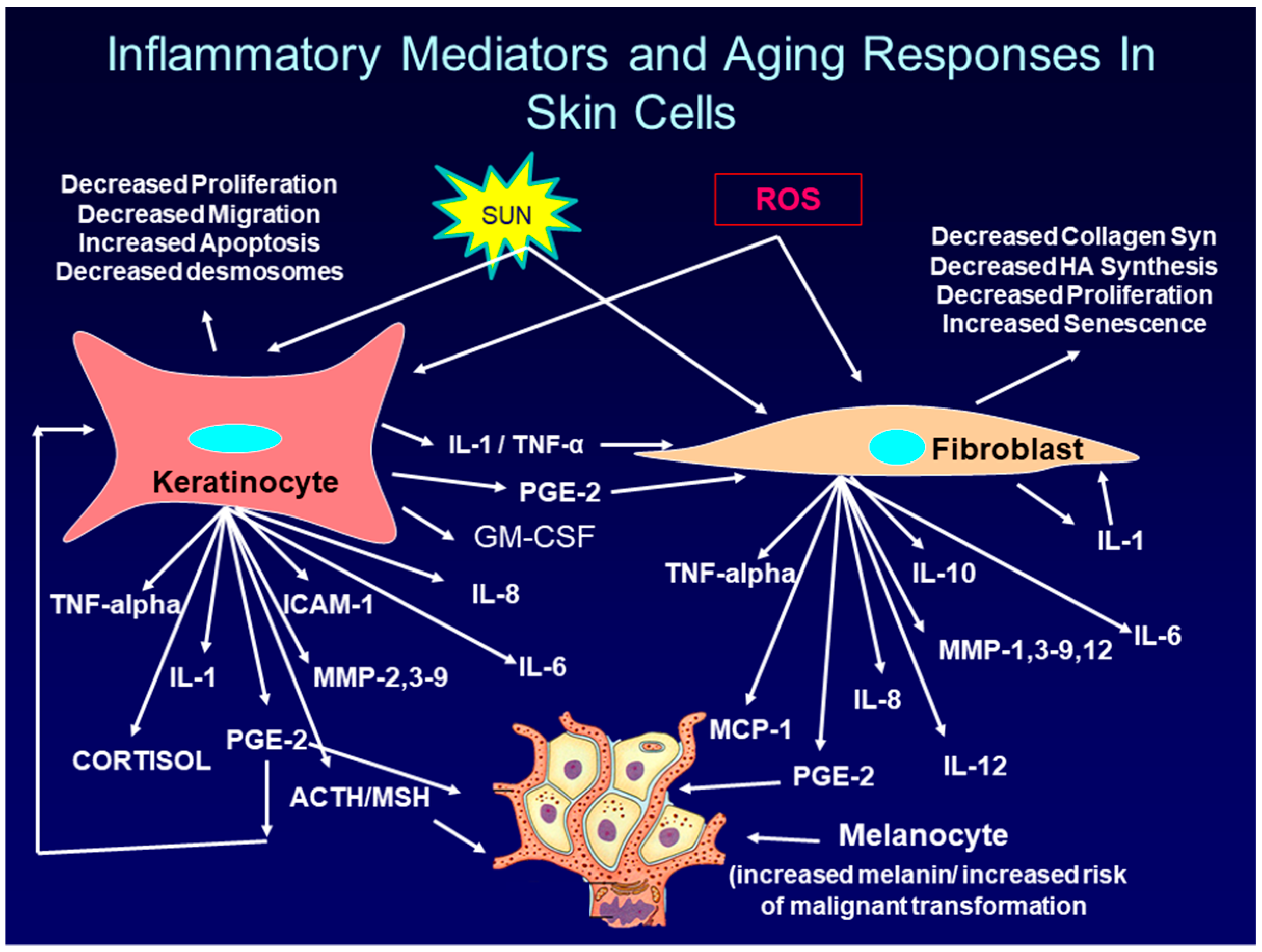
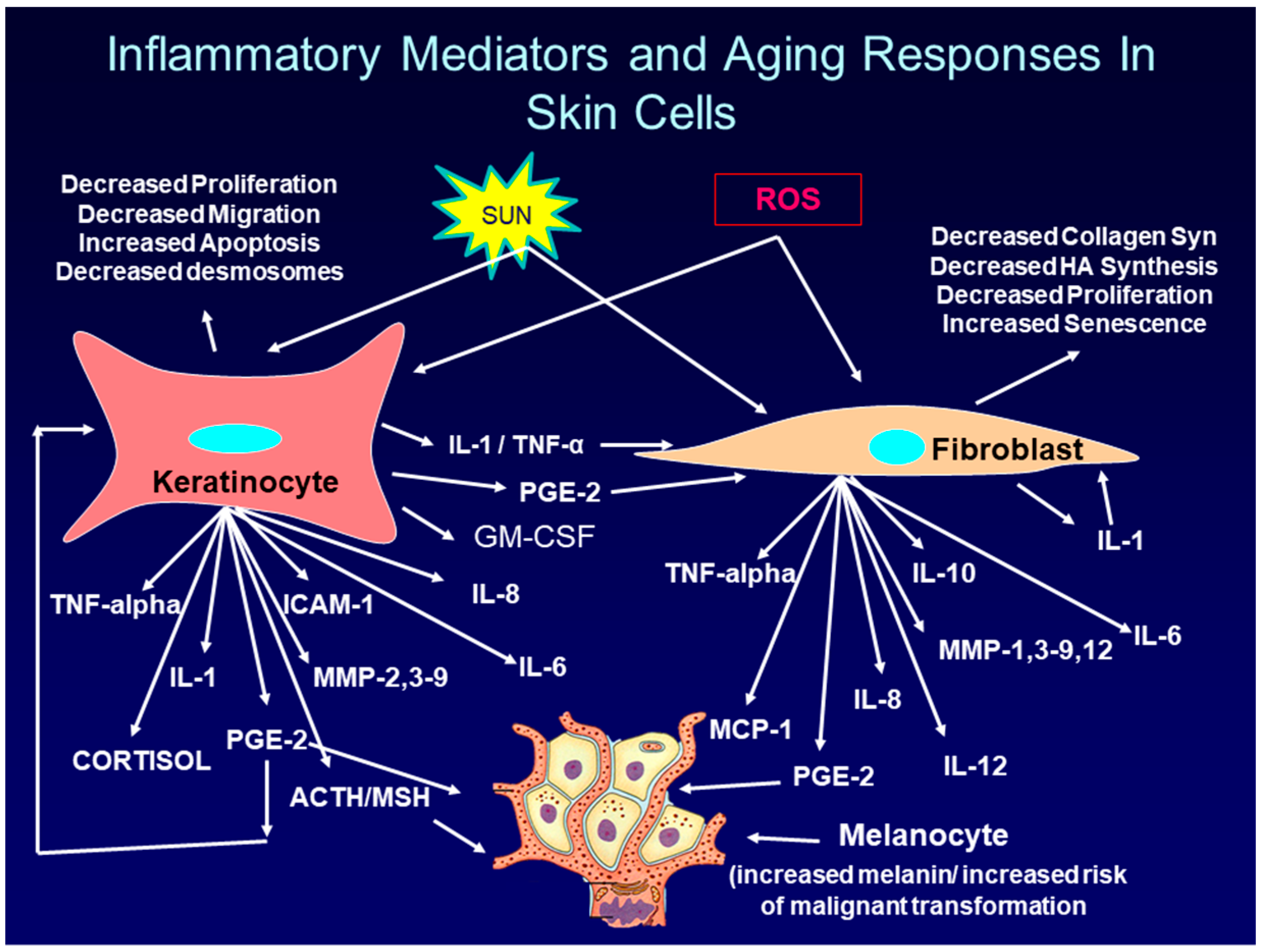
:1. Introduction: Overview of Intrinsic and Extrinsic Skin Aging
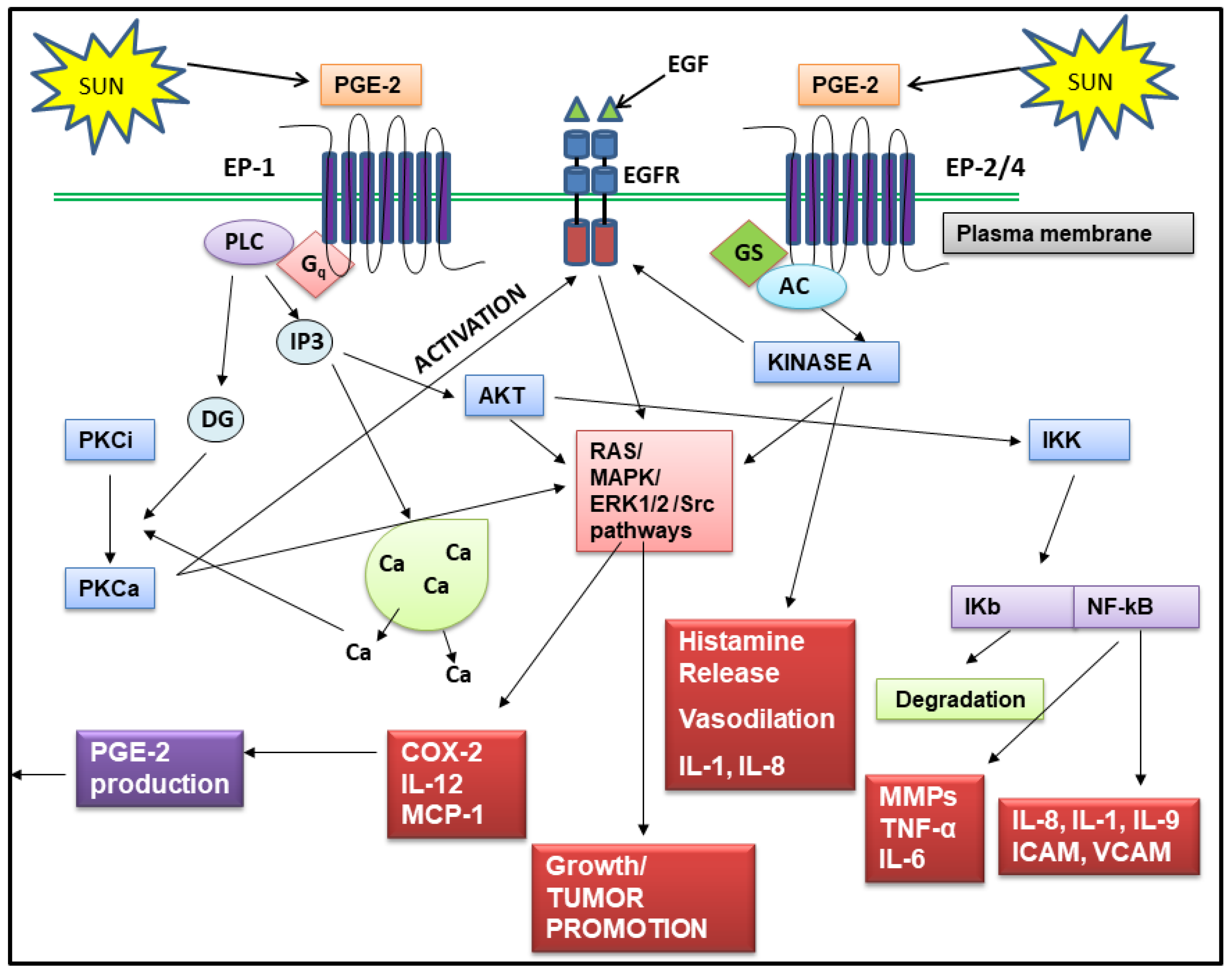
2. The Role of PGE-2 in Skin Aging and Skin Cancer
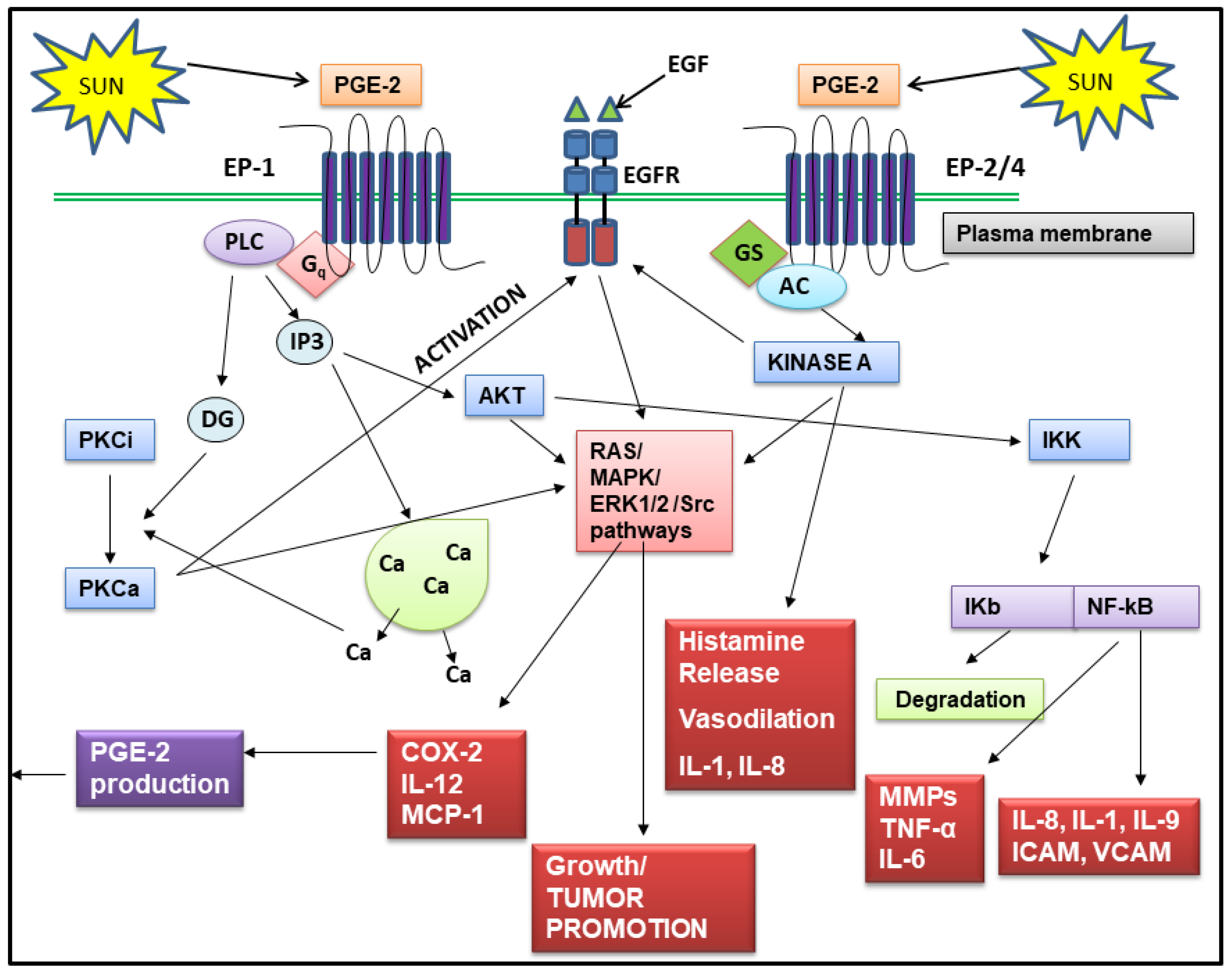
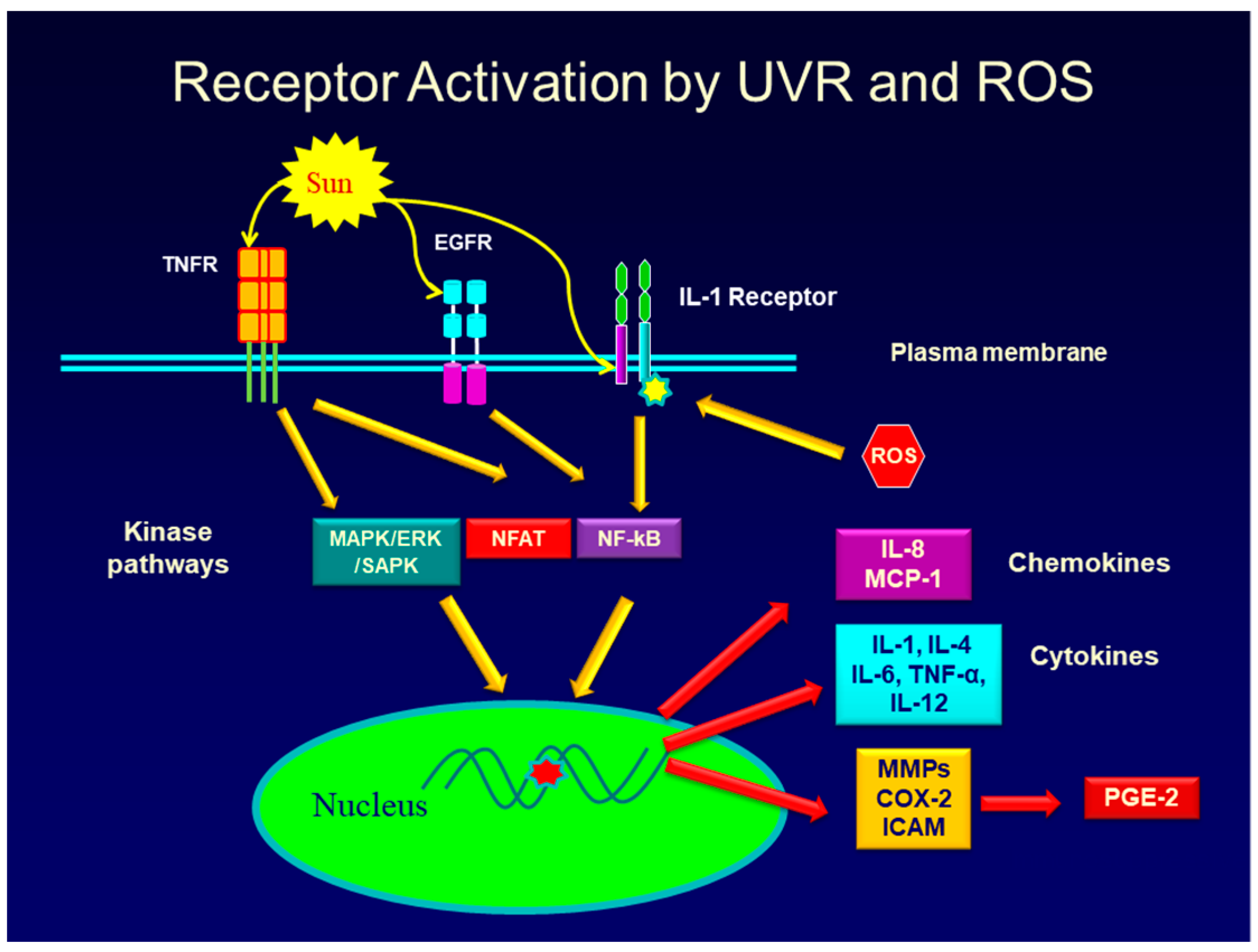
2.1. Regulation of PGE-2 Production in Skin
2.2. PGE-2 Signals Through Four Receptors
- increased scarring during wound healing [120],
2.3. Effects of PGE-2 on Skin Cancer
- Treatment of mice either orally or topically with COX-2 inhibitors, indomethacin, diclofenac, or with the selective COX-2 inhibitor, celecoxib, during UVB induced carcinogenesis, prevented the development of tumors by 85%. Further, celecoxib caused regression of pre-existing tumors [158,159,160,161,162,163].
- Irradiation of hairless mice with a UVA sunlamp, similar to those used in tanning beds, resulted in skin cancers being present in 90% of the animals. The tumors were assessed to be squamous cell carcinoma and COX-2 levels in these tumors was elevated. The tumor-bearing mice were then divided into two groups and one group was fed a diet containing celecoxib. After 2.5 months, those animals treated with celecoxib had 50% fewer tumors than the control group [124,158,160,161,163,164].
- Treatment of mice with a prostaglandin EP1 receptor antagonist, reduced the development of tumors after UVB treatment [104]. In transgenic mice that over-expressed the EP1 receptor, treatment with the chemical carcinogen, DMBA (7,12-dimethyl-benz[a]anthracene), produced a nine fold increase in skin carcinomas compared to wild-type mice [125].
- In melanoma patients, 93% to 95% of the tumors expressed COX-2, while no benign nevi were positive for COX-2. Treatment of human melanoma cell cultures with a specific COX-2 inhibitor prevented migration and invasion of melanoma cells suggesting that lowering PGE-2 may reduce metastasis [121,169]. Further, there was a significant correlation between COX-2 expression and disease-specific survival [169].
- The use of the COX-2 inhibitor, celecoxib, along with a PKC inhibitor, reduced melanoma metastasis in mice injected with melanoma cells [170].
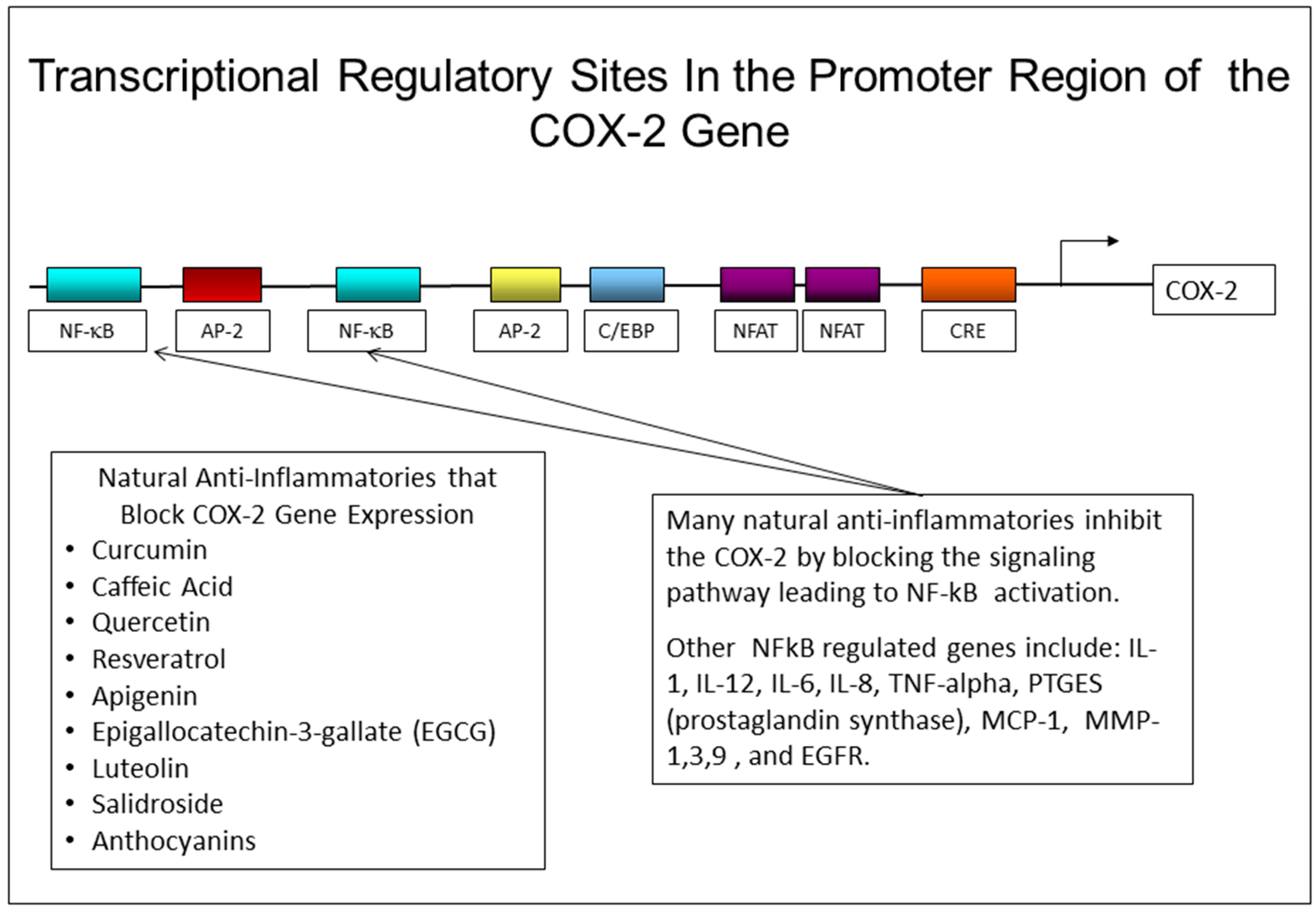
2.4. Effects Inhibition of PGE-2 Production by NSAIDs and Natural Compounds
- Parthenolide, a sesquiterpene lactone found in the commonly used herb, Feverfew, has been shown to block NF-kB regulated genes and inhibits the expression of MMP-1 [190,191]. In addition, by blocking NF-kB activation, parthenolide can inhibit COX-2 gene expression [192]. There is also evidence from the UVB mouse model, that oral treatment of irradiated mice with parthenolide can reduce the number of papillomas induced by UVB treatment [193].
- Resveratrol, a polyphenol found in red wine, has considerable anti-inflammatory activity, and can block the increase in COX-2 and PGE-2 levels induced by UVR, as well as by pollutants [194]. Resveratrol can also block the increase in COX-2 in mice treated with chemical carcinogens [195,196]. It has been shown to block MMP-1 expression [194,197], and NF-kB activation [198]. In addition, the analogue of resveratrol, pterostilbene, can inhibit COX-2 in mouse skin and prevent tumor formation in DMBA-treated mice by inhibiting both NF-kB and MAPK signaling pathways [199,200].
- Green tea, and more specifically, Epigallocatechin-3-gallate (EGCG) from green tea, has been widely studied for its antioxidant, anti-inflammatory, anti-cancer, and anti-aging properties, but the data is conflicting and the overall benefits of EGCG are not clear. In human studies, topical application of EGCG before UV irradiation decreased the UVR induction of ROS and inhibited the infiltration of monocytes into the skin [201]. However, oral supplements containing green tea were ineffective in protecting skin from UVR damage [188]. Studies with human keratinocytes treated with airborne pollutants (PM10), showed that EGCG blocked the pollutant induced increase in NADH oxidases, IL-1, TNF-alpha, IL-8, and MMP-1 [202]. In other studies, EGCG reduced the UVR-induced increase in COX-2 in human keratinocytes, but had the opposite effect on human fibroblast cultures, where it increased the expression of both COX-2 and MMP-1 [203]. In studies with skin equivalent cell culture models, EGCG was found to decrease MMP-1 expression and increase TIMP-1, although the effect was not pronounced [204]. Finally, in studies with human fibroblast cell cultures, EGCG exerted a pronounced down-regulation (40% reduction) in collagen I synthesis [205]. These contradictory findings suggest that EGCG may not be an appropriate ingredient to use in topical products designed to address photoaged or chronically aged skin.
- Aloe Vera extracts are found in many topical products that make marketing claims for helping to reduce inflammation and discomfort from sunburns and inflammatory skin problems. However, there are actually very few scientific studies that have assessed the ability of aloe to suppress inflammatory mediator production in skin. Aloe Vera contains two polyphenolic compounds, Aloin and Aloe-emodin, that are thought to account for the anti-inflammatory effects of the plant. In studies with mouse macrophage cultures induced by LPS (lipopolysaccharide) to produce inflammatory mediators, Aloe-emodin, but not Aloin was found to block COX-2 mRNA expression [206]. In vivo studies using the hairless mouse model showed that a topical Aloe Vera gel extract could reduce the level of MMPs expressed in skin in response to UVB treatment. The effect of this extract on COX-2 levels in UVB-treated skin was not examined in this study [207].
- Other natural compounds that are found in dietary supplements as well as in skin care products, and which have been shown to block either cytokine mediated or UVB-induced COX-2 expression in fibroblasts and/or keratinocytes are: apigenin [186,208], licochalcone [209], salidroside [210], eupafolin [211], CoQ10 [212], delphinidin (from grapes, cranberries) [213], quercetin [187], orange peel extract [214], ferulic acid [215], and luteolin [216].
2.5. Developing Effective Topical Products to Block PGE-2 in Skin
- Anti-inflammatory compounds should block the production and/or action of PGE-2 as well as other skin aging inflammatory mediators, such as IL-1 and TNF-alpha.
- Compounds must have a molecular weight less than 500 Daltons, which is the upper limit for penetration through the stratum corneum.
- For skin penetration, compounds should have a logP (partition coefficient) value between 1 and 3.
- Compounds must be formulated at a high enough concentration to provide bioactivity when applied topically.
- Compounds must be chemically stable when formulated into topical products.
- Formulations should deliver compounds into the skin at a rate that provides benefits (e.g., blocking MMPs) for many hours after a single application.
- Compounds should be colorless and odorless.
3. Conclusions
- If consumers in their teens start using products that lower PGE-2 levels, will their skin look noticeably younger as they age, even if they continue to spend time in the sun, and
- If, at an early age, consumers start using skin care and sun care products that block PGE-2, will they ever get skin cancer or even develop actinic keratosis, even in their later years, and even if they spend time outdoors?
Funding
Conflicts of Interest
References
- Debacq-Chainiaux, F.; Leduc, C.; Verbeke, A.; Toussaint, O. UV, stress and aging. Dermato-Endocrinology 2012, 4, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef]
- Tobin, D.J. Introduction to skin aging. J. Tissue Viability 2017, 26, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Characteristics of the Aging Skin. Adv. Wound Care 2013, 2, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Weihermann, A.C.; Lorencini, M.; Brohem, C.A.; de Carvalho, C.M. Elastin structure and its involvement in skin photoageing. Int. J. Cosmet. Sci. 2017, 39, 241–247. [Google Scholar] [CrossRef]
- Seleit, I.; Bakry, O.A.; El Repey, H.S.; Ali, R. Intrinsic versus Extrinsic Aging: A Histopathological, Morphometric and Immunohistochemical Study of Estrogen Receptor β and Androgen Receptor. Skin Pharmacol. Physiol. 2016, 29, 178–189. [Google Scholar] [CrossRef]
- Poljšak, B.; Dahmane, R.G.; Godić, A. Intrinsic skin aging: The role of oxidative stress. Acta Dermatovenerol. Alp. Pannonica Adriat. 2012, 21, 33–36. [Google Scholar]
- Fang, J.-Y.; Wang, P.-W.; Huang, C.-H.; Chen, M.-H.; Wu, Y.-R.; Pan, T.-L. Skin aging caused by intrinsic or extrinsic processes characterized with functional proteomics. Proteomics 2016, 16, 2718–2731. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Uitto, J. The role of elastin and collagen in cutaneous aging: Intrinsic aging versus photoexposure. J. Drugs Dermatol. 2008, 7, s12–s16. [Google Scholar]
- Sárdy, M. Role of Matrix Metalloproteinases in Skin Ageing. Connect. Tissue Res. 2009, 50, 132–138. [Google Scholar] [CrossRef]
- Rinnerthaler, M.; Bischof, J.; Streubel, M.; Trost, A.; Richter, K. Oxidative Stress in Aging Human Skin. Biomolecules 2015, 5, 545–589. [Google Scholar] [CrossRef] [Green Version]
- Kossodo, S.; Wong, W.-R.; Simon, G.; Kochevar, I.E. Effects of UVR and UVR-induced Cytokines on Production of Extracellular Matrix Proteins and Proteases by Dermal Fibroblasts Cultured in Collagen Gels. Photochem. Photobiol. 2004, 79, 86–93. [Google Scholar] [CrossRef]
- Sauvaigo, S.; Bonnet-Duquennoy, M.; Odin, F.; Hazane-Puch, F.; Lachmann, N.; Bonté, F.; Kurfürst, R.; Favier, A. DNA repair capacities of cutaneous fibroblasts: Effect of sun exposure, age and smoking on response to an acute oxidative stress. Br. J. Dermatol. 2007, 157, 26–32. [Google Scholar] [CrossRef]
- Kim, C.; Ryu, H.-C.; Kim, J.-H. Low-dose UVB irradiation stimulates matrix metalloproteinase-1 expression via a BLT2-linked pathway in HaCaT cells. Exp. Mol. Med. 2010, 42, 833–841. [Google Scholar] [CrossRef]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-Degrading Metalloproteinases in Photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, M.-B.; Yun, J.G.; Hwang, J.K. Protective Effects of Standardized Siegesbeckia glabrescens Extract and Its Active Compound Kirenol against UVB-Induced Photoaging through Inhibition of MAPK/NF-κB Pathways. J. Microbiol. Biotechnol. 2017, 27, 242–250. [Google Scholar] [CrossRef]
- Tewari, A.; Grys, K.; Kollet, J.; Sarkany, R.; Young, A.R. Upregulation of MMP12 and Its Activity by UVA1 in Human Skin: Potential Implications for Photoaging. J. Investig. Dermatol. 2014, 134, 2598–2609. [Google Scholar] [CrossRef] [Green Version]
- Dai, G.; Freudenberger, T.; Zipper, P.; Melchior, A.; Grether-Beck, S.; Rabausch, B.; de Groot, J.; Twarock, S.; Hanenberg, H.; Homey, B.; et al. Chronic ultraviolet B irradiation causes loss of hyaluronic acid from mouse dermis because of down-regulation of hyaluronic acid synthases. Am. J. Pathol. 2007, 171, 1451–1461. [Google Scholar] [CrossRef]
- Shin, J.-E.; Oh, J.-H.; Kim, Y.K.; Jung, J.-Y.; Chung, J.H. Transcriptional Regulation of Proteoglycans and Glycosaminoglycan Chain-synthesizing Glycosyltransferases by UV Irradiation in Cultured Human Dermal Fibroblasts. J. Korean Med. Sci. 2011, 26, 417–424. [Google Scholar] [CrossRef]
- Kurdykowski, S.; Mine, S.; Bardey, V.; Danoux, L.; Jeanmaire, C.; Pauly, G.; Brabencova, E.; Wegrowski, Y.; Maquart, F.X. Ultraviolet-B Irradiation Induces Differential Regulations of Hyaluronidase Expression and Activity in Normal Human Keratinocytes: Photochemistry and Photobiology. Photochem. Photobiol. 2011, 87, 1105–1112. [Google Scholar] [CrossRef]
- Oh, J.-H.; Kim, Y.K.; Jung, J.-Y.; Shin, J.; Kim, K.H.; Cho, K.H.; Eun, H.C.; Chung, J.H. Intrinsic aging- and photoaging-dependent level changes of glycosaminoglycans and their correlation with water content in human skin. J. Dermatol. Sci. 2011, 62, 192–201. [Google Scholar] [CrossRef]
- Glady, A.; Tanaka, M.; Moniaga, C.S.; Yasui, M.; Hara-Chikuma, M. Involvement of NADPH oxidase 1 in UVB-induced cell signaling and cytotoxicity in human keratinocytes. Biochem. Biophys. Rep. 2018, 14, 7–15. [Google Scholar] [CrossRef]
- Kammeyer, A.; Luiten, R.M. Oxidation events and skin aging. Ageing Res. Rev. 2015, 21, 16–29. [Google Scholar] [CrossRef]
- Kozina, L.S.; Borzova, I.V.; Arutiunov, V.A.; Ryzhak, G.A. Role of oxidative stress in skin aging. Adv. Gerontol. 2013, 3, 18–22. [Google Scholar] [CrossRef]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef]
- Valencia, A.; Kochevar, I.E. Nox1-Based NADPH Oxidase Is the Major Source of UVA-Induced Reactive Oxygen Species in Human Keratinocytes. J. Investig. Dermatol. 2008, 128, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Beak, S.M.; Lee, Y.S.; Kim, J.-A. NADPH oxidase and cyclooxygenase mediate the ultraviolet B-induced generation of reactive oxygen species and activation of nuclear factor-κB in HaCaT human keratinocytes. Biochimie 2004, 86, 425–429. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, Y.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J. Cell. Biochem. 2009, 107, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.P.; Wu, J.X.; Fan, Y.; Adamson, E.D. UV activates growth factor receptors via reactive oxygen intermediates. J. Cell Biol. 1996, 133, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Tober, K.L.; Thomas-Ahner, J.M.; Kusewitt, D.F.; Oberyszyn, T.M. Effects of UVB on E Prostanoid Receptor Expression in Murine Skin. J. Investig. Dermatol. 2007, 127, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Soontrapa, K.; Honda, T.; Sakata, D.; Yao, C.; Hirata, T.; Hori, S.; Matsuoka, T.; Kita, Y.; Shimizu, T.; Kabashima, K.; et al. Prostaglandin E2-prostoglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc. Natl. Acad. Sci. USA 2011, 108, 6668–6673. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFκB and TNFα signal transduction pathways. Arch. Dermatol. Res. 2010, 302, 5–17. [Google Scholar] [CrossRef]
- Wan, Y.S.; Wang, Z.Q.; Voorhees, J.; Fisher, G. EGF receptor crosstalks with cytokine receptors leading to the activation of c-Jun kinase in response to UV irradiation in human keratinocytes. Cell. Signal. 2001, 13, 139–144. [Google Scholar] [CrossRef]
- Blanton, R.A.; Kupper, T.S.; McDougall, J.K.; Dower, S. Regulation of interleukin 1 and its receptor in human keratinocytes. Proc. Natl. Acad. Sci. USA 1989, 86, 1273–1277. [Google Scholar] [CrossRef]
- López-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Pérez-Plasencia, C.; Alvarez-Sánchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci. 2012, 13, 142–172. [Google Scholar] [CrossRef]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar] [CrossRef]
- Peus, D.; Meves, A.; Vasa, R.A.; Beyerle, A.; O’Brien, T.; Pittelkow, M.R. H2O2 is required for UVB-induced EGF receptor and downstream signaling pathway activation. Free Radic. Biol. Med. 1999, 27, 1197–1202. [Google Scholar] [CrossRef]
- Madson, J.G.; Hansen, L.A. Multiple mechanisms of Erbb2 action after ultraviolet irradiation of the skin. Mol. Carcinog. 2007, 46, 624–628. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Chaiprasongsuk, A.; Lohakul, J.; Soontrapa, K.; Sampattavanich, S.; Akarasereenont, P.; Panich, U. Activation of Nrf2 Reduces UVA-Mediated MMP-1 Upregulation via MAPK/AP-1 Signaling Cascades: The Photoprotective Effects of Sulforaphane and Hispidulin. J. Pharmacol. Exp. Ther. 2017, 360, 388–398. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Yuan, W.; Mori, Y.; Varga, J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene 2000, 19, 3546–3555. [Google Scholar] [CrossRef]
- Chung, K.Y.; Agarwal, A.; Uitto, J.; Mauviel, A. An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J. Biol. Chem. 1996, 271, 3272–3278. [Google Scholar] [CrossRef]
- Marrot, L. Pollution and Sun Exposure: A Deleterious Synergy. Mechanisms and Opportunities for Skin Protection. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Crisan, M.; Taulescu, M.; Crisan, D.; Cosgarea, R.; Parvu, A.; Cãtoi, C.; Drugan, T. Expression of Advanced Glycation End-Products on Sun-Exposed and Non-Exposed Cutaneous Sites during the Ageing Process in Humans. PLoS ONE 2013, 8, e75003. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Martien, S.; Pluquet, O.; Vercamer, C.; Malaquin, N.; Martin, N.; Gosselin, K.; Pourtier, A.; Abbadie, C. Cellular senescence involves an intracrine prostaglandin E2 pathway in human fibroblasts. Biochim. Biophys. Acta 2013, 1831, 1217–1227. [Google Scholar] [CrossRef]
- Fossel, M. Cell senescence in human aging and disease. Ann. N. Y. Acad. Sci. 2002, 959, 14–23. [Google Scholar] [CrossRef]
- Toutfaire, M.; Bauwens, E.; Debacq-Chainiaux, F. The impact of cellular senescence in skin ageing: A notion of mosaic and therapeutic strategies. Biochem. Pharmacol. 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Bi, Z.; Chu, W.; Wan, Y. IL-1 receptor antagonist attenuates MAP kinase/AP-1 activation and MMP1 expression in UVA-irradiated human fibroblasts induced by culture medium from UVB-irradiated human skin keratinocytes. Int. J. Mol. Med. 2005, 16, 1117–1124. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef]
- Kasza, A. IL-1 and EGF regulate expression of genes important in inflammation and cancer. Cytokine 2013, 62, 22–33. [Google Scholar] [CrossRef]
- Trefzer, U.; Brockhaus, M.; Lötscher, H.; Parlow, F.; Budnik, A.; Grewe, M.; Christoph, H.; Kapp, A.; Schöpf, E.; Luger, T.A. The 55-kD tumor necrosis factor receptor on human keratinocytes is regulated by tumor necrosis factor-alpha and by ultraviolet B radiation. J. Clin. Investig. 1993, 92, 462–470. [Google Scholar] [CrossRef]
- Takii, T.; Akahoshi, T.; Kato, K.; Hayashi, H.; Marunouchi, T.; Onozaki, K. Interleukin-1 up-regulates transcription of its own receptor in a human fibroblast cell line TIG-1: Role of endogenous PGE2 and cAMP. Eur. J. Immunol. 1992, 22, 1221–1227. [Google Scholar] [CrossRef]
- El-Abaseri, T.B.; Hammiller, B.; Repertinger, S.K.; Hansen, L.A. The Epidermal Growth Factor Receptor Increases Cytokine Production and Cutaneous Inflammation in Response to Ultraviolet Irradiation. ISRN Dermatol. 2013, 2013, 848705. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef]
- Li, Y.; Lei, D.; Swindell, W.R.; Xia, W.; Weng, S.; Fu, J.; Worthen, C.A.; Okubo, T.; Johnston, A.; Gudjonsson, J.E.; et al. Age-Associated Increase in Skin Fibroblast–Derived Prostaglandin E2 Contributes to Reduced Collagen Levels in Elderly Human Skin. J. Investig. Dermatol. 2015, 135, 2181–2188. [Google Scholar] [CrossRef]
- Habib, M.A.; Salem, S.A.M.; Hakim, S.A.; Shalan, Y.A.M. Comparative immunohistochemical assessment of cutaneous cyclooxygenase-2 enzyme expression in chronological aging and photoaging: COX-2 in chronological aging and photoaging. Photodermatol. Photoimmunol. Photomed. 2014, 30, 43–51. [Google Scholar] [CrossRef]
- Surowiak, P.; Gansukh, T.; Donizy, P.; Halon, A.; Rybak, Z. Increase in cyclooxygenase-2 (COX-2) expression in keratinocytes and dermal fibroblasts in photoaged skin. J. Cosmet. Dermatol. 2014, 13, 195–201. [Google Scholar] [CrossRef]
- Ashida, M.; Bito, T.; Budiyanto, A.; Ichihashi, M.; Ueda, M. Involvement of EGF receptor activation in the induction of cyclooxygenase-2 in HaCaT keratinocytes after UVB. Exp. Dermatol. 2003, 12, 445–452. [Google Scholar] [CrossRef]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Bachelor, M.A.; Cooper, S.J.; Sikorski, E.T.; Bowden, G.T. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol. Cancer Res. MCR 2005, 3, 90–99. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef] [Green Version]
- Flockhart, R.J.; Diffey, B.L.; Farr, P.M.; Lloyd, J.; Reynolds, N.J. NFAT regulates induction of COX-2 and apoptosis of keratinocytes in response to ultraviolet radiation exposure. FASEB J. 2008, 22, 4218–4227. [Google Scholar] [CrossRef] [Green Version]
- Båge, T.; Lindberg, J.; Lundeberg, J.; Modéer, T.; Yucel-Lindberg, T. Signal pathways JNK and NF-κB, identified by global gene expression profiling, are involved in regulation of TNFα-induced mPGES-1 and COX-2 expression in gingival fibroblasts. BMC Genomics 2010, 11, 241. [Google Scholar] [CrossRef]
- Wen, K.-C.; Fan, P.-C.; Tsai, S.-Y.; Shih, I.-C.; Chiang, H.-M. Ixora parviflora Protects against UVB-Induced Photoaging by Inhibiting the Expression of MMPs, MAP Kinases, and COX-2 and by Promoting Type I Procollagen Synthesis. Evid. Based Complement. Alternat. Med. 2012, 2012, 417346. [Google Scholar] [CrossRef]
- Mazière, C.; Morlière, P.; Louandre, C.; Conte, M.-A.; Gomilla, C.; Santus, R.; Antonicelli, F.; Hornebeck, W.; Mazière, J.-C. Low UVA doses activate the transcription factor NFAT in human fibroblasts by a calcium-calcineurin pathway. Free Radic. Biol. Med. 2005, 39, 1629–1637. [Google Scholar] [CrossRef]
- Zucali, J.R.; Dinarello, C.A.; Oblon, D.J.; Gross, M.A.; Anderson, L.; Weiner, R.S. Interleukin 1 stimulates fibroblasts to produce granulocyte-macrophage colony-stimulating activity and prostaglandin E2. J. Clin. Investig. 1986, 77, 1857–1863. [Google Scholar] [CrossRef]
- Kida, Y.; Kobayashi, M.; Suzuki, T.; Takeshita, A.; Okamatsu, Y.; Hanazawa, S.; Yasui, T.; Hasegawa, K. Interleukin-1 stimulates cytokines, prostaglandin E and matrix metalloproteinase-1 production via activation of MAPK/AP-1 and NF-κB in human gingival fibroblasts. Cytokine 2005, 29, 159–168. [Google Scholar] [CrossRef]
- Bagga, D.; Wang, L.; Farias-Eisner, R.; Glaspy, J.A.; Reddy, S.T. Differential effects of prostaglandin derived from -6 and -3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 1751–1756. [Google Scholar] [CrossRef] [Green Version]
- Ansari, K.M.; Sung, Y.M.; He, G.; Fischer, S.M. Prostaglandin receptor EP2 is responsible for cyclooxygenase-2 induction by prostaglandin E2 in mouse skin. Carcinogenesis 2007, 28, 2063–2068. [Google Scholar] [CrossRef]
- Kim, B.-H.; Oh, I.; Kim, J.-H.; Jeon, J.-E.; Jeon, B.; Shin, J.; Kim, T.-Y. Anti-inflammatory activity of compounds isolated from Astragalus sinicus L. in cytokine-induced keratinocytes and skin. Exp. Mol. Med. 2014, 46, e87. [Google Scholar] [CrossRef]
- Seo, S.-H.; Jeong, G.-S. Fisetin inhibits TNF-α-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte HaCaT cells through PI3K/AKT/Nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef]
- Maldve, R.E.; Kim, Y.; Muga, S.J.; Fischer, S.M. Prostaglandin E(2) regulation of cyclooxygenase expression in keratinocytes is mediated via cyclic nucleotide-linked prostaglandin receptors. J. Lipid Res. 2000, 41, 873–881. [Google Scholar]
- Black, A.T.; Gray, J.P.; Shakarjian, M.P.; Mishin, V.; Laskin, D.L.; Heck, D.E.; Laskin, J.D. UVB light upregulates prostaglandin synthases and prostaglandin receptors in mouse keratinocytes. Toxicol. Appl. Pharmacol. 2008, 232, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Rijken, F.; Kiekens, R.C.M.; Bruijnzeel, P.L.B. Skin-infiltrating neutrophils following exposure to solar-simulated radiation could play an important role in photoageing of human skin. Br. J. Dermatol. 2005, 152, 321–328. [Google Scholar] [CrossRef]
- Su, Y.; Richmond, A. Chemokine Regulation of Neutrophil Infiltration of Skin Wounds. Adv. Wound Care 2015, 4, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, M.; Doble, M. Transcriptional Regulation of mPGES1 in Cancer: An Alternative Approach to Drug Discovery? Curr. Drug Targets 2017, 18, 119–131. [Google Scholar] [CrossRef]
- Páramo, J.A.; Rodríguez, J.A.; Beloqui, O.; Orbe, J. Monocyte cyclooxygenase-2 activity: A new therapeutic target for atherosclerosis? Curr. Drug Targets Cardiovasc. Haematol. Disord. 2005, 5, 303–311. [Google Scholar] [CrossRef]
- Cheng, S.-E.; Luo, S.-F.; Jou, M.-J.; Lin, C.-C.; Kou, Y.R.; Lee, I.-T.; Hsieh, H.-L.; Yang, C.-M. Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-kappaB in human tracheal smooth muscle cells. Free Radic. Biol. Med. 2009, 46, 948–960. [Google Scholar] [CrossRef]
- Zhou, X.; Li, D.; Resnick, M.B.; Wands, J.; Cao, W. NADPH Oxidase NOX5-S and Nuclear Factor B1 Mediate Acid-Induced Microsomal Prostaglandin E Synthase-1 Expression in Barrett’s Esophageal Adenocarcinoma Cells. Mol. Pharmacol. 2013, 83, 978–990. [Google Scholar] [CrossRef]
- Akitake, Y.; Nakatani, Y.; Kamei, D.; Hosokawa, M.; Akatsu, H.; Uematsu, S.; Akira, S.; Kudo, I.; Hara, S.; Takahashi, M. Microsomal prostaglandin E synthase-1 is induced in alzheimer’s disease and its deletion mitigates alzheimer’s disease-like pathology in a mouse model: mPGES-1 in Alzheimer’s Disease. J. Neurosci. Res. 2013, 91, 909–919. [Google Scholar] [CrossRef]
- Kojima, F.; Kato, S.; Kawai, S. Prostaglandin E synthase in the pathophysiology of arthritis. Fundam. Clin. Pharmacol. 2005, 19, 255–261. [Google Scholar] [CrossRef]
- Koeberle, A.; Laufer, S.A.; Werz, O. Design and Development of Microsomal Prostaglandin E2 Synthase-1 Inhibitors: Challenges and Future Directions. J. Med. Chem. 2016, 59, 5970–5986. [Google Scholar] [CrossRef]
- Shekfeh, S.; Çalışkan, B.; Fischer, K.; Yalçın, T.; Garscha, U.; Werz, O.; Banoglu, E. A Multi-step Virtual Screening Protocol for the Identification of Novel Non-acidic Microsomal Prostaglandin E2 Synthase-1 (mPGES-1) Inhibitors. ChemMedChem 2018. [Google Scholar] [CrossRef]
- Chen, X.; Gresham, A.; Morrison, A.; Pentland, A.P. Oxidative stress mediates synthesis of cytosolic phospholipase A2 after UVB injury. Biochim. Biophys. Acta 1996, 1299, 23–33. [Google Scholar] [CrossRef]
- Gresham, A.; Masferrer, J.; Chen, X.; Leal-Khouri, S.; Pentland, A.P. Increased synthesis of high-molecular-weight cPLA2 mediates early UV-induced PGE2 in human skin. Am. J. Physiol.-Cell Physiol. 1996, 270, C1037–C1050. [Google Scholar] [CrossRef]
- Lin, C.-C.; Lin, W.-N.; Cho, R.-L.; Wang, C.; Hsiao, L.-D.; Yang, C.-M. TNF-α-Induced cPLA2 Expression via NADPH Oxidase/Reactive Oxygen Species-Dependent NF-κB Cascade on Human Pulmonary Alveolar Epithelial Cells. Front. Pharmacol. 2016, 7, 447. [Google Scholar] [CrossRef]
- Sjursen, W.; Brekke, O.L.; Johansen, B. Secretory and cytosolic phospholipase A(2)regulate the long-term cytokine-induced eicosanoid production in human keratinocytes. Cytokine 2000, 12, 1189–1194. [Google Scholar] [CrossRef]
- Lee, C.-W.; Lee, I.-T.; Lin, C.-C.; Lee, H.-C.; Lin, W.-N.; Yang, C.-M. Activation and induction of cytosolic phospholipase A2 by IL-1Î2 in human tracheal smooth muscle cells: Role of MAPKs/p300 and NF-ÎoB. J. Cell. Biochem. 2010, 109, 1045–1056. [Google Scholar]
- Luo, S.-F.; Lin, C.-C.; Chen, H.-C.; Lin, W.-N.; Lee, I.-T.; Lee, C.-W.; Hsiao, L.-D.; Yang, C.-M. Involvement of MAPKs, NF-κB and p300 co-activator in IL-1β-induced cytosolic phospholipase A2 expression in canine tracheal smooth muscle cells. Toxicol. Appl. Pharmacol. 2008, 232, 396–407. [Google Scholar] [CrossRef]
- Rundhaug, J.E.; Simper, M.S.; Surh, I.; Fischer, S.M. The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev. 2011, 30, 465–480. [Google Scholar] [CrossRef]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.J. G protein-coupled receptor structures, molecular associations, and modes of regulation. Ann. N. Y. Acad. Sci. 2008, 1144, 1–5. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Tober, K.L.; Wilgus, T.A.; Kusewitt, D.F.; Thomas-Ahner, J.M.; Maruyama, T.; Oberyszyn, T.M. Importance of the EP1 Receptor in Cutaneous UVB-Induced Inflammation and Tumor Development. J. Investig. Dermatol. 2006, 126, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Konger, R.L.; Malaviya, R.; Pentland, A.P. Growth regulation of primary human keratinocytes by prostaglandin E receptor EP2 and EP3 subtypes. Biochim. Biophys. Acta 1998, 1401, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.M.; He, G.; Hwang, D.H.; Fischer, S.M. Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. Oncogene 2006, 25, 5507–5516. [Google Scholar] [CrossRef]
- Konger, R.L.; Brouxhon, S.; Partillo, S.; VanBuskirk, J.; Pentland, A.P. The EP3 receptor stimulates ceramide and diacylglycerol release and inhibits growth of primary keratinocytes. Exp. Dermatol. 2005, 14, 914–922. [Google Scholar] [CrossRef]
- Amano, H.; Ito, Y.; Suzuki, T.; Kato, S.; Matsui, Y.; Ogawa, F.; Murata, T.; Sugimoto, Y.; Senior, R.; Kitasato, H.; et al. Roles of a prostaglandin E-type receptor, EP3, in upregulation of matrix metalloproteinase-9 and vascular endothelial growth factor during enhancement of tumor metastasis. Cancer Sci. 2009, 100, 2318–2324. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; An, S.; Goetzl, E.J. Regulation of expression of matrix metalloproteinase-9 in early human T cells of the HSB.2 cultured line by the EP3 subtype of prostaglandin E2 receptor. J. Biol. Chem. 1996, 271, 27744–27750. [Google Scholar] [CrossRef]
- Yokoyama, U.; Iwatsubo, K.; Umemura, M.; Fujita, T.; Ishikawa, Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol. Rev. 2013, 65, 1010–1052. [Google Scholar] [CrossRef] [Green Version]
- Konya, V.; Marsche, G.; Schuligoi, R.; Heinemann, A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013, 138, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Tandara, A.A.; Mustoe, T.A. MMP- and TIMP-secretion by human cutaneous keratinocytes and fibroblasts--impact of coculture and hydration. J. Plast. Reconstr. Aesthet. Surg. 2011, 64, 108–116. [Google Scholar] [CrossRef]
- Yen, J.-H.; Khayrullina, T.; Ganea, D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood 2008, 111, 260–270. [Google Scholar] [CrossRef]
- Steenport, M.; Khan, K.M.F.; Du, B.; Barnhard, S.E.; Dannenberg, A.J.; Falcone, D.J. Matrix Metalloproteinase (MMP)-1 and MMP-3 Induce Macrophage MMP-9: Evidence for the Role of TNF- and Cyclooxygenase-2. J. Immunol. 2009, 183, 8119–8127. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Kang, J.S.; Kim, H.N.; Jung, D.J.; Kim, J.E.; Mun, G.H.; Kim, Y.S.; Cho, D.; Shin, D.H.; Hwang, Y.-I.; Lee, W.J. Regulation of UVB-Induced IL-8 and MCP-1 Production in Skin Keratinocytes by Increasing Vitamin C Uptake via the Redistribution of SVCT-1 from the Cytosol to the Membrane. J. Investig. Dermatol. 2007, 127, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Conti, P.; DiGioacchino, M. MCP-1 and RANTES are mediators of acute and chronic inflammation. Allergy Asthma Proc. 2001, 22, 133–137. [Google Scholar] [CrossRef]
- Varga, J.; Diaz-Perez, A.; Rosenbloom, J.; Jimenez, S.A. PGE2 causes a coordinate decrease in the steady state levels of fibronectin and types I and III procollagen mRNAs in normal human dermal fibroblasts. Biochem. Biophys. Res. Commun. 1987, 147, 1282–1288. [Google Scholar] [CrossRef]
- Yang, H.H.; Kim, C.; Jung, B.; Kim, K.S.; Kim, J.-R. Involvement of IGF binding protein 5 in prostaglandin E2-induced cellular senescence in human fibroblasts. Biogerontology 2011, 12, 239–252. [Google Scholar] [CrossRef]
- Romana-Souza, B.; dos Santos, J.S.; Bandeira, L.G.; Monte-Alto-Costa, A. Selective inhibition of COX-2 improves cutaneous wound healing of pressure ulcers in mice through reduction of iNOS expression. Life Sci. 2016, 153, 82–92. [Google Scholar] [CrossRef]
- Denkert, C.; Köbel, M.; Berger, S.; Siegert, A.; Leclere, A.; Trefzer, U.; Hauptmann, S. Expression of cyclooxygenase 2 in human malignant melanoma. Cancer Res. 2001, 61, 303–308. [Google Scholar]
- Thompson, E.J.; Gupta, A.; Vielhauer, G.A.; Regan, J.W.; Bowden, G.T. The growth of malignant keratinocytes depends on signaling through the PGE2 receptor EP1. Neoplasia 2001, 3, 402–410. [Google Scholar] [CrossRef]
- Bachelor, M.A.; Bowden, G.T. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin. Cancer Biol. 2004, 14, 131–138. [Google Scholar] [CrossRef]
- Pentland, A.P.; Schoggins, J.W.; Scott, G.A.; Khan, K.N.; Han, R. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis 1999, 20, 1939–1944. [Google Scholar] [CrossRef] [Green Version]
- Surh, I.; Rundhaug, J.; Pavone, A.; Mikulec, C.; Abel, E.; Fischer, S.M. Upregulation of the EP1 receptor for prostaglandin E2 promotes skin tumor progression. Mol. Carcinog. 2011, 50, 458–468. [Google Scholar] [CrossRef]
- Rijken, F.; Bruijnzeel, P.L.B. The Pathogenesis of Photoaging: The Role of Neutrophils and Neutrophil-Derived Enzymes. J. Investig. Dermatol. Symp. Proc. 2009, 14, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Wong, W.R.; Kossodo, S.; Kochevar, I.E. Influence of cytokines on matrix metalloproteinases produced by fibroblasts cultured in monolayer and collagen gels. J. Formos. Med. Assoc./Taiwan Yi Zhi 2001, 100, 377–382. [Google Scholar]
- Zhao, J.; Shu, B.; Chen, L.; Tang, J.; Zhang, L.; Xie, J.; Liu, X.; Xu, Y.; Qi, S. Prostaglandin E2 inhibits collagen synthesis in dermal fibroblasts and prevents hypertrophic scar formation in vivo. Exp. Dermatol. 2016, 25, 604–610. [Google Scholar] [CrossRef]
- Thampatty, B.P.; Li, H.; Im, H.-J.; Wang, J.H.-C. EP4 receptor regulates collagen type-I, MMP-1, and MMP-3 gene expression in human tendon fibroblasts in response to IL-1 beta treatment. Gene 2007, 386, 154–161. [Google Scholar] [CrossRef]
- Hayashi, T.; Nishihira, J.; Koyama, Y.; Sasaki, S.; Yamamoto, Y. Decreased Prostaglandin E2 Production by Inflammatory Cytokine and Lower Expression of EP2 Receptor Result in Increased Collagen Synthesis in Keloid Fibroblasts. J. Investig. Dermatol. 2006, 126, 990–997. [Google Scholar] [CrossRef]
- Liu, X. Fibrotic Lung Fibroblasts Show Blunted Inhibition by cAMP Due to Deficient cAMP Response Element-Binding Protein Phosphorylation. J. Pharmacol. Exp. Ther. 2005, 315, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Inoue, T.; Higaki, M.; Mizushima, Y. Cyclooxygenase inhibitors enhance the production of tissue inhibitor-1 of metalloproteinases (TIMP-1) and pro-matrix metalloproteinase 1 (proMMP-1) in human rheumatoid synovial fibroblasts. Inflamm. Res. 1997, 46, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Banu, S.K.; Subbarao, T.; Starzinski-Powitz, A.; Arosh, J.A. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 inhibits invasion of human immortalized endometriotic epithelial and stromal cells through suppression of metalloproteinases. Mol. Cell. Endocrinol. 2011, 332, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Itahana, Y.; Dimri, G.P. Colorimetric detection of senescence-associated β galactosidase. Methods Mol. Biol. 2013, 965, 143–156. [Google Scholar]
- Han, J.H.; Roh, M.S.; Park, C.-H.; Park, K.C.; Cho, K.H.; Kim, K.H.; Eun, H.C.; Chung, J.H. Selective COX-2 inhibitor, NS-398, inhibits the replicative senescence of cultured dermal fibroblasts. Mech. Ageing Dev. 2004, 125, 359–366. [Google Scholar] [CrossRef]
- Goorochurn, R.; Viennet, C.; Granger, C.; Fanian, F.; Varin-Blank, N.; Roy, C.L.; Humbert, P. Biological processes in solar lentigo: Insights brought by experimental models. Exp. Dermatol. 2016, 25, 174–177. [Google Scholar] [CrossRef]
- Lee, B.W.; Schwartz, R.A.; Janniger, C.K. Melasma. G. Ital. Dermatol. E Venereol. Organo Uff. Soc. Ital. Dermatol. E Sifilogr. 2017, 152, 36–45. [Google Scholar]
- Schalka, S. New data on hyperpigmentation disorders. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymińska, A. Skin melanocytes: Biology and development. Postepy Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Imokawa, G.; Miyagishi, M.; Yada, Y. Endothelin-1 as a new melanogen: Coordinated expression of its gene and the tyrosinase gene in UVB-exposed human epidermis. J. Investig. Dermatol. 1995, 105, 32–37. [Google Scholar] [CrossRef]
- Starner, R.J.; McClelland, L.; Abdel-Malek, Z.; Fricke, A.; Scott, G. PGE2 is a UVR-inducible autocrine factor for human melanocytes that stimulates tyrosinase activation. Exp. Dermatol. 2010, 19, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Pernet, I.; Mayoux, C.; Trompezinski, S.; Schmitt, D.; Viac, J. Modulation of endothelin-1 in normal human keratinocytes by UVA1/B radiations, prostaglandin E2 and peptidase inhibitors. Exp. Dermatol. 2000, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Gledhill, K.; Rhodes, L.E.; Brownrigg, M.; Haylett, A.K.; Masoodi, M.; Thody, A.J.; Nicolaou, A.; Tobin, D.J. Prostaglandin-E2 is produced by adult human epidermal melanocytes in response to UVB in a melanogenesis-independent manner. Pigment Cell Melanoma Res. 2010, 23, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Fricke, A.; Fender, A.; McClelland, L.; Jacobs, S. Prostaglandin E2 regulates melanocyte dendrite formation through activation of PKCzeta. Exp. Cell Res. 2007, 313, 3840–3850. [Google Scholar] [CrossRef]
- Ma, H.-J.; Ma, H.-Y.; Yang, Y.; Li, P.-C.; Zi, S.-X.; Jia, C.-Y.; Chen, R. α-Melanocyte stimulating hormone (MSH) and prostaglandin E2 (PGE2) drive melanosome transfer by promoting filopodia delivery and shedding spheroid granules: Evidences from atomic force microscopy observation. J. Dermatol. Sci. 2014, 76, 222–230. [Google Scholar] [CrossRef]
- Scott, G.; Leopardi, S.; Printup, S.; Malhi, N.; Seiberg, M.; Lapoint, R. Proteinase-activated receptor-2 stimulates prostaglandin production in keratinocytes: Analysis of prostaglandin receptors on human melanocytes and effects of PGE2 and PGF2alpha on melanocyte dendricity. J. Investig. Dermatol. 2004, 122, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Le Clair, M.Z.; Cockburn, M.G. Tanning bed use and melanoma: Establishing risk and improving prevention interventions. Prev. Med. Rep. 2016, 3, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, A.J.; Subbaramaiah, K. Targeting cyclooxygenase-2 in human neoplasia: Rationale and promise. Cancer Cell 2003, 4, 431–436. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.M.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef]
- Kochel, T.J.; Goloubeva, O.G.; Fulton, A.M. Upregulation of Cyclooxygenase-2/Prostaglandin E2 (COX-2/PGE2) Pathway Member Multiple Drug Resistance-Associated Protein 4 (MRP4) and Downregulation of Prostaglandin Transporter (PGT) and 15-Prostaglandin Dehydrogenase (15-PGDH) in Triple-Negative Breast Cancer. Breast Cancer Basic Clin. Res. 2016, 10, 61–70. [Google Scholar]
- Müller-Decker, K. Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: Pharmacological, genetic, and clinical evidence. Cancer Metastasis Rev. 2011, 30, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, L.-Q.; Wei, W. Prostanoids receptors signaling in different diseases/cancers progression. J. Recept. Signal Transduct. Res. 2013, 33, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, T.; Higashi, Y.; Kanzaki, T. Inhibitory effects of 9-cis-retinoic acid and pyrrolidinedithiocarbamate on cyclooxygenase (COX)-2 expression and cell growth in human skin squamous carcinoma cells. Cancer Lett. 2000, 161, 177–183. [Google Scholar] [CrossRef]
- An, K.P.; Athar, M.; Tang, X.; Katiyar, S.K.; Russo, J.; Beech, J.; Aszterbaum, M.; Kopelovich, L.; Epstein, E.H.; Mukhtar, H.; et al. Cyclooxygenase-2 expression in murine and human nonmelanoma skin cancers: Implications for therapeutic approaches. Photochem. Photobiol. 2002, 76, 73–80. [Google Scholar] [CrossRef]
- Fischer, S.M. Is cyclooxygenase-2 important in skin carcinogenesis? J. Environ. Pathol. Toxicol. Oncol. 2002, 21, 183–191. [Google Scholar] [CrossRef]
- Zhan, H.; Zheng, H. The role of topical cyclo-oxygenase-2 inhibitors in skin cancer: Treatment and prevention. Am. J. Clin. Dermatol. 2007, 8, 195–200. [Google Scholar] [CrossRef]
- Elmets, C.A.; Ledet, J.J.; Athar, M. Cyclooxygenases: Mediators of UV-Induced Skin Cancer and Potential Targets for Prevention. J. Investig. Dermatol. 2014, 134, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.M.; Lo, H.H.; Gordon, G.B.; Seibert, K.; Kelloff, G.; Lubet, R.A.; Conti, C.J. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol. Carcinog. 1999, 25, 231–240. [Google Scholar] [CrossRef]
- Burns, E.M.; Tober, K.L.; Riggenbach, J.A.; Schick, J.S.; Lamping, K.N.; Kusewitt, D.F.; Young, G.S.; Oberyszyn, T.M. Preventative topical diclofenac treatment differentially decreases tumor burden in male and female Skh-1 mice in a model of UVB-induced cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Koki, A.T.; Zweifel, B.S.; Kusewitt, D.F.; Rubal, P.A.; Oberyszyn, T.M. Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topical celecoxib treatment. Mol. Carcinog. 2003, 38, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Koki, A.T.; Zweifel, B.S.; Rubal, P.A.; Oberyszyn, T.M. Chemotherapeutic efficacy of topical celecoxib in a murine model of ultraviolet light B-induced skin cancer. Mol. Carcinog. 2003, 38, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E.; Fischer, S.M. Cyclo-oxygenase-2 Plays a Critical Role in UV-induced Skin Carcinogenesis. Photochem. Photobiol. 2008, 84, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Decker, K.; Fürstenberger, G. The cyclooxygenase-2-mediated prostaglandin signaling is causally related to epithelial carcinogenesis. Mol. Carcinog. 2007, 46, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Tiano, H.F.; Loftin, C.D.; Akunda, J.; Lee, C.A.; Spalding, J.; Sessoms, A.; Dunson, D.B.; Rogan, E.G.; Morham, S.G.; Smart, R.C.; et al. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002, 62, 3395–3401. [Google Scholar] [PubMed]
- Buckman, S.Y.; Gresham, A.; Hale, P.; Hruza, G.; Anast, J.; Masferrer, J.; Pentland, A.P. COX-2 expression is induced by UVB exposure in human skin: Implications for the development of skin cancer. Carcinogenesis 1998, 19, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.R.; Siegelin, M.D.; Rompel, R.; Enk, A.H.; Gaiser, T. COX-2 expression in malignant melanoma: A novel prognostic marker? Melanoma Res. 2009, 19, 8–16. [Google Scholar] [CrossRef]
- Zhou, P.; Qin, J.; Li, Y.; Li, G.; Wang, Y.; Zhang, N.; Chen, P.; Li, C. Combination therapy of PKCζ and COX-2 inhibitors synergistically suppress melanoma metastasis. J. Exp. Clin. Cancer Res. 2017, 36, 115. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Aszterbaum, M.; Athar, M.; Barsanti, F.; Cappola, C.; Estevez, N.; Hebert, J.; Hwang, J.; Khaimskiy, Y.; Kim, A.; et al. Basal Cell Carcinoma Chemoprevention with Nonsteroidal Anti-inflammatory Drugs in Genetically Predisposed PTCH1+/− Humans and Mice. Cancer Prev. Res. (Phila. Pa.) 2010, 3, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Ross, M.S.; Parrett, M.L.; Oberyszyn, T.M. Topical application of a selective cyclooxygenase inhibitor suppresses UVB mediated cutaneous inflammation. Prostaglandins Other Lipid Mediat. 2000, 62, 367–384. [Google Scholar] [CrossRef]
- Hatamipour, M.; Johnston, T.P.; Sahebkar, A. One Molecule, Many Targets and Numerous Effects: The Pleiotropy of Curcumin Lies in its Chemical Structure. Curr. Pharm. Des. 2018, 24, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, R.; Shi, H.; Li, X.; Li, Y.; Taha, A.; Xu, C. Protective effect of curcumin against ultraviolet A irradiation-induced photoaging in human dermal fibroblasts. Mol. Med. Rep. 2018, 17, 7227–7237. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-R.; Parnell, L.D.; Ordovas, J.M.; Lai, C.-Q. Curcumin and aging. BioFactors 2013, 39, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Jobin, C.; Bradham, C.A.; Russo, M.P.; Juma, B.; Narula, A.S.; Brenner, D.A.; Sartor, R.B. Curcumin blocks cytokine-mediated NF-kappa B activation and proinflammatory gene expression by inhibiting inhibitory factor I-kappa B kinase activity. J. Immunol. 1999, 163, 3474–3483. [Google Scholar]
- Shishodia, S. Molecular mechanisms of curcumin action: Gene expression. BioFactors 2013, 39, 37–55. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef]
- Wang, J.; Ma, J.; Gu, J.-H.; Wang, F.-Y.; Shang, X.-S.; Tao, H.-R.; Wang, X. Regulation of type II collagen, matrix metalloproteinase-13 and cell proliferation by interleukin-1β is mediated by curcumin via inhibition of NF-κB signaling in rat chondrocytes. Mol. Med. Rep. 2017, 16, 1837–1845. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.L.; Forsey, R.J.; Blades, T.J.; Barratt, M.E.; Parmar, P.; Powell, J.R. Antioxidants may contribute in the fight against ageing: An in vitro model. Mech. Ageing Dev. 2000, 121, 217–230. [Google Scholar] [CrossRef]
- Palmer, D.M.; Kitchin, J.S. Oxidative damage, skin aging, antioxidants and a novel antioxidant rating system. J. Drugs Dermatol. 2010, 9, 11–15. [Google Scholar] [PubMed]
- Pandel, R.; Poljšak, B.; Godic, A.; Dahmane, R. Skin photoaging and the role of antioxidants in its prevention. ISRN Dermatol. 2013, 2013, 930164. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-W.; Park, K.; Kweon, G.R.; Jang, B.-C.; Baek, W.-K.; Suh, M.-H.; Kim, C.-W.; Lee, K.-S.; Suh, S.-I. Curcumin inhibits the expression of COX-2 in UVB-irradiated human keratinocytes (HaCaT) by inhibiting activation of AP-1: p38 MAP kinase and JNK as potential upstream targets. Exp. Mol. Med. 2005, 37, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.M.; Lee, K.W.; Jung, S.K.; Lee, E.J.; Heo, Y.-S.; Bode, A.M.; Lubet, R.A.; Lee, H.J.; Dong, Z. Kaempferol inhibits UVB-induced COX-2 expression by suppressing Src kinase activity. Biochem. Pharmacol. 2010, 80, 2042–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Van Dross, R.T.; Abu-Yousif, A.; Morrison, A.R.; Pelling, J.C. Apigenin Prevents UVB-Induced Cyclooxygenase 2 Expression: Coupled mRNA Stabilization and Translational Inhibition. Mol. Cell. Biol. 2007, 27, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.-S.; Lee, E.-G.; Jeon, H.-S.; Chae, H.-J.; Park, S.J.; Lee, Y.C.; Yoo, W.-H. Quercetin Inhibits IL-1β-Induced Proliferation and Production of MMPs, COX-2, and PGE2 by Rheumatoid Synovial Fibroblast. Inflammation 2012, 35, 1585–1594. [Google Scholar] [CrossRef]
- Farrar, M.D.; Nicolaou, A.; Clarke, K.A.; Mason, S.; Massey, K.A.; Dew, T.P.; Watson, R.E.; Williamson, G.; Rhodes, L.E. A randomized controlled trial of green tea catechins in protection against ultraviolet radiation–induced cutaneous inflammation. Am. J. Clin. Nutr. 2015, 102, 608–615. [Google Scholar] [CrossRef]
- Yarla, N.S.; Bishayee, A.; Sethi, G.; Reddanna, P.; Kalle, A.M.; Dhananjaya, B.L.; Dowluru, K.S.V.G.K.; Chintala, R.; Duddukuri, G.R. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin. Cancer Biol. 2016, 40–41, 48–81. [Google Scholar] [CrossRef]
- Nam, N.-H. Naturally occurring NF-kappaB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945–951. [Google Scholar] [CrossRef]
- Tanaka, K.; Asamitsu, K.; Uranishi, H.; Iddamalgoda, A.; Ito, K.; Kojima, H.; Okamoto, T. Protecting skin photoaging by NF-kappaB inhibitor. Curr. Drug Metab. 2010, 11, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Fischer, N.H.; Jang, B.C.; Tak, H.; Kim, J.K.; Lee, W. Inhibition of the expression of inducible cyclooxygenase and proinflammatory cytokines by sesquiterpene lactones in macrophages correlates with the inhibition of MAP kinases. Biochem. Biophys. Res. Commun. 1996, 226, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Won, Y.-K. Chemopreventive activity of parthenolide against UVB-induced skin cancer and its mechanisms. Carcinogenesis 2004, 25, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Hsu, L.-F.; Lee, C.-W.; Chiang, Y.-C.; Lee, M.-H.; How, J.-M.; Wu, C.-M.; Huang, C.-L.; Lee, I.-T. Resveratrol inhibits urban particulate matter-induced COX-2/PGE2 release in human fibroblast-like synoviocytes via the inhibition of activation of NADPH oxidase/ROS/NF-κB. Int. J. Biochem. Cell Biol. 2017, 88, 113–123. [Google Scholar] [CrossRef]
- Kowalczyk, M.C.; Junco, J.J.; Kowalczyk, P.; Tolstykh, O.; Hanausek, M.; Slaga, T.J.; Walaszek, Z. Effects of combined phytochemicals on skin tumorigenesis in SENCAR mice. Int. J. Oncol. 2013, 43, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.-J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.-H.; Kim, S.Y. Resveratrol-Enriched Rice Attenuates UVB-ROS-Induced Skin Aging via Downregulation of Inflammatory Cascades. Oxid. Med. Cell. Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef]
- Ren, Z.; Wang, L.; Cui, J.; Huoc, Z.; Xue, J.; Cui, H.; Mao, Q.; Yang, R. Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Pharmazie 2013, 68, 689–694. [Google Scholar]
- Tsai, M.-L.; Lai, C.-S.; Chang, Y.-H.; Chen, W.-J.; Ho, C.-T.; Pan, M.-H. Pterostilbene, a natural analogue of resveratrol, potently inhibits 7,12-dimethylbenz[a]anthracene (DMBA)/12-O-tetradecanoylphorbol-13-acetate (TPA)-induced mouse skin carcinogenesis. Food Funct. 2012, 3, 1185–1194. [Google Scholar] [CrossRef]
- Cichocki, M.; Paluszczak, J.; Szaefer, H.; Piechowiak, A.; Rimando, A.M.; Baer-Dubowska, W. Pterostilbene is equally potent as resveratrol in inhibiting 12-O-tetradecanoylphorbol-13-acetate activated NFkappaB, AP-1, COX-2, and iNOS in mouse epidermis. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S62–S70. [Google Scholar]
- Katiyar, S.K.; Afaq, F.; Perez, A.; Mukhtar, H. Green tea polyphenol (-)-epigallocatechin-3-gallate treatment of human skin inhibits ultraviolet radiation-induced oxidative stress. Carcinogenesis 2001, 22, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, J.K.; Lee, J.; Kim, Y.M.; Boo, Y.C. Punicalagin and (–)-Epigallocatechin-3-Gallate Rescue Cell Viability and Attenuate Inflammatory Responses of Human Epidermal Keratinocytes Exposed to Airborne Particulate Matter PM10. Skin Pharmacol. Physiol. 2018, 31, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Soriani, M.; Rice-Evans, C.; Tyrrell, R.M. Modulation of the UVA activation of haem oxygenase, collagenase and cyclooxygenase gene expression by epigallocatechin in human skin cells. FEBS Lett. 1998, 439, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Chung, J.H.; Cho, K.H. The effects of epigallocatechin-3-gallate on extracellular matrix metabolism. J. Dermatol. Sci. 2005, 40, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Dooley, A.; Shi-Wen, X.; Aden, N.; Tranah, T.; Desai, N.; Denton, C.P.; Abraham, D.J.; Bruckdorfer, R. Modulation of collagen type I, fibronectin and dermal fibroblast function and activity, in systemic sclerosis by the antioxidant epigallocatechin-3-gallate. Rheumatology 2010, 49, 2024–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.-Y.; Kwon, H.-J.; Sung, M.-K. Evaluation of aloin and aloe-emodin as anti-inflammatory agents in aloe by using murine macrophages. Biosci. Biotechnol. Biochem. 2009, 73, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Misawa, E.; Tanaka, M.; Saito, M.; Nabeshima, K.; Yao, R.; Yamauchi, K.; Abe, F.; Yamamoto, Y.; Furukawa, F. Protective effects of Aloe sterols against UVB-induced photoaging in hairless mice. Photodermatol. Photoimmunol. Photomed. 2017, 33, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, A.J.; Soliman, E.; Jenkins, A.; Van Dross, R.T. Apigenin inhibits COX-2, PGE2, and EP1 and also initiates terminal differentiation in the epidermis of tumor bearing mice. Prostaglandins Leukot. Essent. Fat. Acids 2016, 104, 44–53. [Google Scholar] [CrossRef]
- Furuhashi, I.; Iwata, S.; Sato, T.; Inoue, H.; Shibata, S. Inhibition by licochalcone A, a novel flavonoid isolated from liquorice root, of IL-1β-induced PGE2 production in human skin fibroblasts. J. Pharm. Pharmacol. 2005, 57, 1661–1666. [Google Scholar] [CrossRef]
- Wu, D.; Yuan, P.; Ke, C.; Xiong, H.; Chen, J.; Guo, J.; Lu, M.; Ding, Y.; Fan, X.; Duan, Q.; et al. Salidroside suppresses solar ultraviolet-induced skin inflammation by targeting cyclooxygenase-2. Oncotarget 2016, 7, 25971–25982. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Lin, Z.-C.; Liang, C.-J.; Yen, F.-L.; Chiang, Y.-C.; Lee, C.-W. Eupafolin inhibits PGE2 production and COX2 expression in LPS-stimulated human dermal fibroblasts by blocking JNK/AP-1 and Nox2/p47phox pathway. Toxicol. Appl. Pharmacol. 2014, 279, 240–251. [Google Scholar] [CrossRef]
- Fuller, B.; Smith, D.; Howerton, A.; Kern, D. Anti-inflammatory effects of CoQ10 and colorless carotenoids. J. Cosmet. Dermatol. 2006, 5, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.Y.; Lee, K.W.; Kim, J.-E.; Jung, S.K.; Kang, N.J.; Hwang, M.K.; Heo, Y.-S.; Bode, A.M.; Dong, Z.; Lee, H.J. Delphinidin suppresses ultraviolet B-induced cyclooxygenases-2 expression through inhibition of MAPKK4 and PI-3 kinase. Carcinogenesis 2009, 30, 1932–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, N.; Fujii, T.; Masaki, H.; Okubo, T.; Shimada, K.; Hashizume, R. Orange peel extract, containing high levels of polymethoxyflavonoid, suppressed UVB-induced COX-2 expression and PGE2 production in HaCaT cells through PPAR-γ activation. Exp. Dermatol. 2014, 23, 18–22. [Google Scholar] [CrossRef]
- Das, U.; Manna, K.; Sinha, M.; Datta, S.; Das, D.K.; Chakraborty, A.; Ghosh, M.; Saha, K.D.; Dey, S. Role of ferulic acid in the amelioration of ionizing radiation induced inflammation: A murine model. PLoS ONE 2014, 9, e97599. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Son, J.E.; Jang, Y.J.; Lee, D.E.; Kang, N.J.; Jung, S.K.; Heo, Y.-S.; Lee, K.W.; Lee, H.J. Luteolin, a novel natural inhibitor of tumor progression locus 2 serine/threonine kinase, inhibits tumor necrosis factor-alpha-induced cyclooxygenase-2 expression in JB6 mouse epidermis cells. J. Pharmacol. Exp. Ther. 2011, 338, 1013–1022. [Google Scholar] [CrossRef]
- Novitskiy, G.; Potter, J.J.; Rennie-Tankersley, L.; Mezey, E. Identification of a Novel NF-κB-binding Site with Regulation of the Murine α2 (I) Collagen Promoter. J. Biol. Chem. 2004, 279, 15639–15644. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kizoulis, M.; Fantasia, J.; Oddos, T.; Bigot, N.; Galera, P.; Tucker-Samaras, S.; Leyden, J.J.; Southall, M.D. 4-Hexyl-1,3-phenylenediol, a nuclear factor-κB inhibitor, improves photodamaged skin and clinical signs of ageing in a double-blinded, randomized controlled trial. Br. J. Dermatol. 2015, 173, 218–226. [Google Scholar] [CrossRef]
- Long, X.; Zeng, X.; Zhang, F.; Wang, X. Influence of quercetin and x-ray on collagen synthesis of cultured human keloid-derived fibroblasts. Chin. Med. Sci. J. 2006, 21, 179–183. [Google Scholar]
- Bos, J.D.; Meinardi, M.M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Korinth, G.; Wellner, T.; Schaller, K.H.; Drexler, H. Potential of the octanol–water partition coefficient (logP) to predict the dermal penetration behaviour of amphiphilic compounds in aqueous solutions. Toxicol. Lett. 2012, 215, 49–53. [Google Scholar] [CrossRef]
- Potts, R.O.; Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 9, 663–669. [Google Scholar] [CrossRef]
- Jäger, R.; Lowery, R.P.; Calvanese, A.V.; Joy, J.M.; Purpura, M.; Wilson, J.M. Comparative absorption of curcumin formulations. Nutr. J. 2014, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.-F.; Rouse, J.J.; Sanderson, F.D.; Meidan, V.; Eccleston, G.M. Validation of a static Franz diffusion cell system for in vitro permeation studies. AAPS PharmSciTech 2010, 11, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Bartosova, L.; Bajgar, J. Transdermal drug delivery in vitro using diffusion cells. Curr. Med. Chem. 2012, 19, 4671–4677. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inflammation Mediated Aging Events in Skin |
| Decreased expression of collagen genes I, III and VII, and particularly collagen-1. |
| Decreased levels of HA and other GAGs. |
| Increased activity of hyaluronidase-1. |
| Increased MMPs causing a loss of collagen and other matrix proteins. |
| Degeneration of the normal elastic fiber network. |
| Increased production of abnormal elastin organization (elastosis). |
| Reduction in lipid synthesis. |
| Loss in energy (ATP) production by mitochondria: increased ROS. |
| Increased influx of immune cells producing inflammatory mediators. |
| Slower keratinocyte and fibroblast cell turnover and cell replacement. |
| Decreased DNA repair of damaged skin cells. |
| Increased senescence of fibroblasts. |
| Increase in Actinic Keratoses and skin cancer. |
| Increase in melanogenesis (hyperpigmentation) and solar lentigines |
| Weak epidermal dermal junction. |
| Increased apoptosis of skin cells. |
| % Inhibition of Inflammatory Mediator Production in UVR or TPA Treated Keratinocytes | ||||
|---|---|---|---|---|
| Mediator → | PGE-2 | TNF-alpha | IL-1 | |
| Compound (100 µM) | Structure ↓ | |||
| Curcumin/Tetrahydrocurcumin | 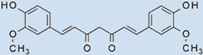 | 100 | 100 | 100 |
| Quercetin |  | 96 | 100 | 88 |
| Luteolin | 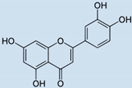 | 69 | 95 | 32 |
| Myricetin | 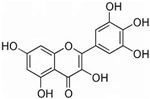 | 5 | 95 | 90 |
| Dihydroeugenol |  | 95 | 65 | 94 |
| Kaempherol | 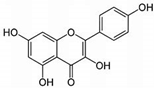 | 100 | 100 | 89 |
| Caffeic Acid | 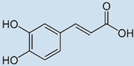 | 31 | 86 | 82 |
| Epigallocatechin Gallate | 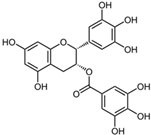 | 42 | 100 | 83 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuller, B. Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories. Cosmetics 2019, 6, 6. https://doi.org/10.3390/cosmetics6010006
Fuller B. Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories. Cosmetics. 2019; 6(1):6. https://doi.org/10.3390/cosmetics6010006
Chicago/Turabian StyleFuller, Bryan. 2019. "Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories" Cosmetics 6, no. 1: 6. https://doi.org/10.3390/cosmetics6010006
APA StyleFuller, B. (2019). Role of PGE-2 and Other Inflammatory Mediators in Skin Aging and Their Inhibition by Topical Natural Anti-Inflammatories. Cosmetics, 6(1), 6. https://doi.org/10.3390/cosmetics6010006