
Uncovering Streptomyces-Derived Compounds as Cosmeceuticals for the Development of Improved Skin Photoprotection Products: An In Silico Approach to Explore Multi-Targeted Agents




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Ligand Selection and Preparation
2.2. Protein Selection and Preparation
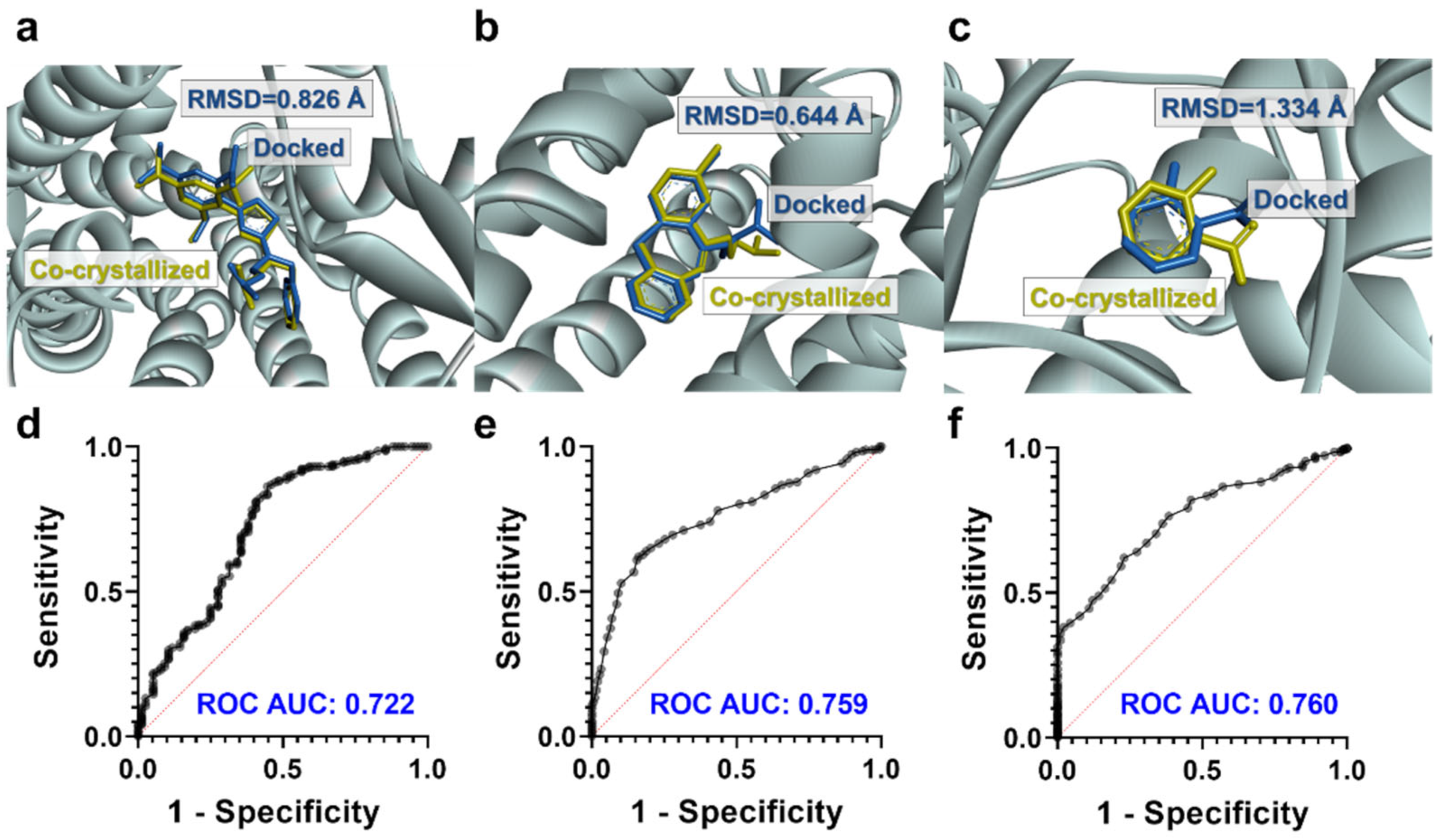
2.3. Molecular Docking Studies
2.4. Quantum Properties of Top-Ranked Compounds and Theoretical Predictions of UV Spectra of A1 and A3
2.5. Molecular Dynamics Simulations
2.6. Data Exploration and Statistical Analysis
3. Results
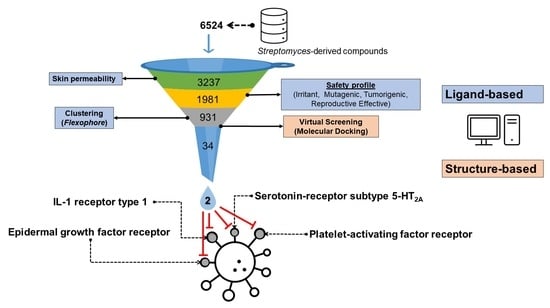
3.1. Filtering of Streptomyces-Derived Compounds
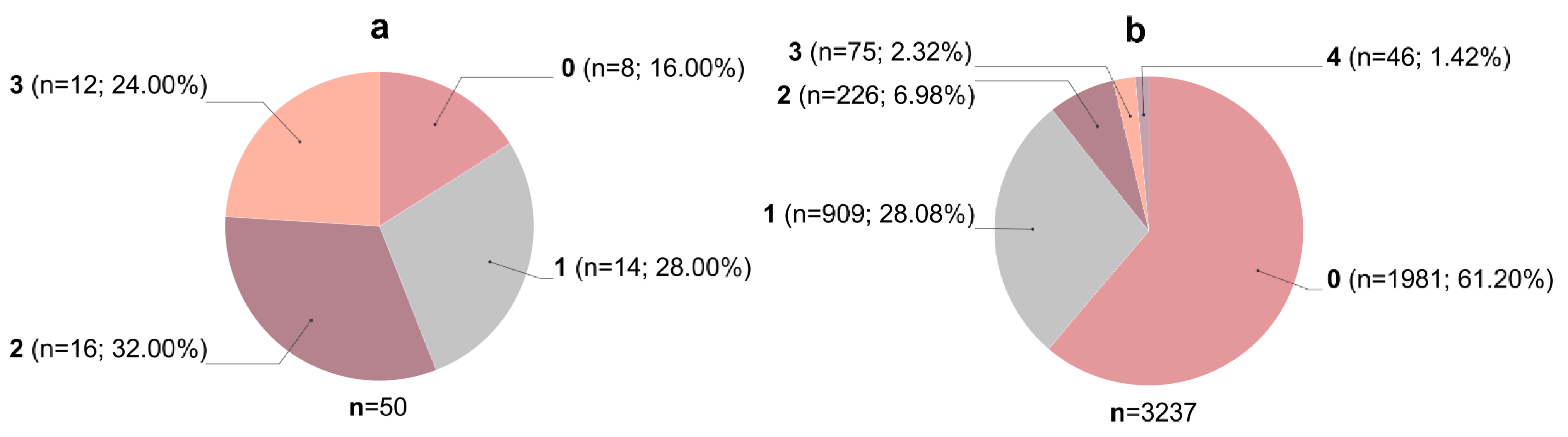
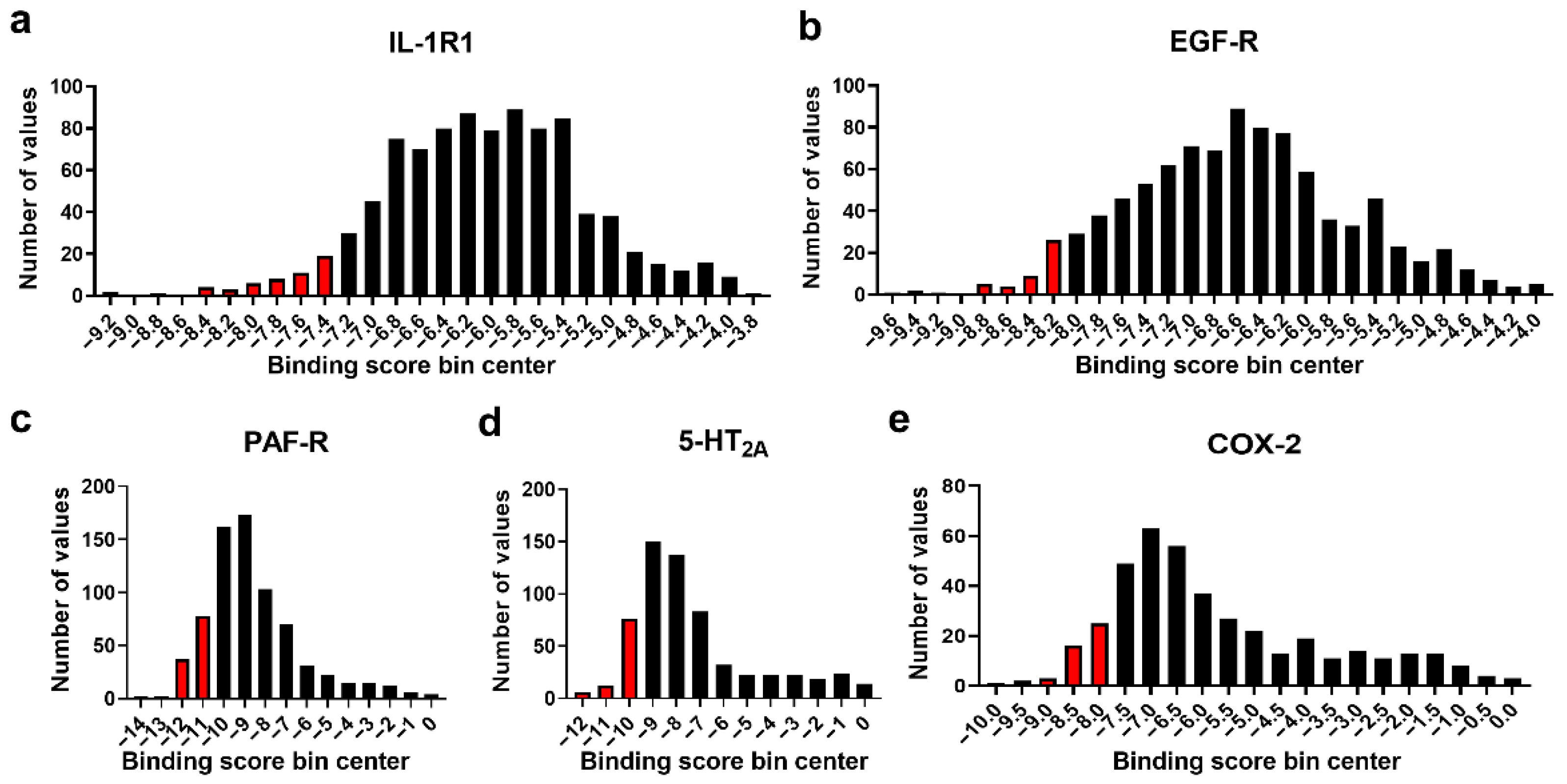
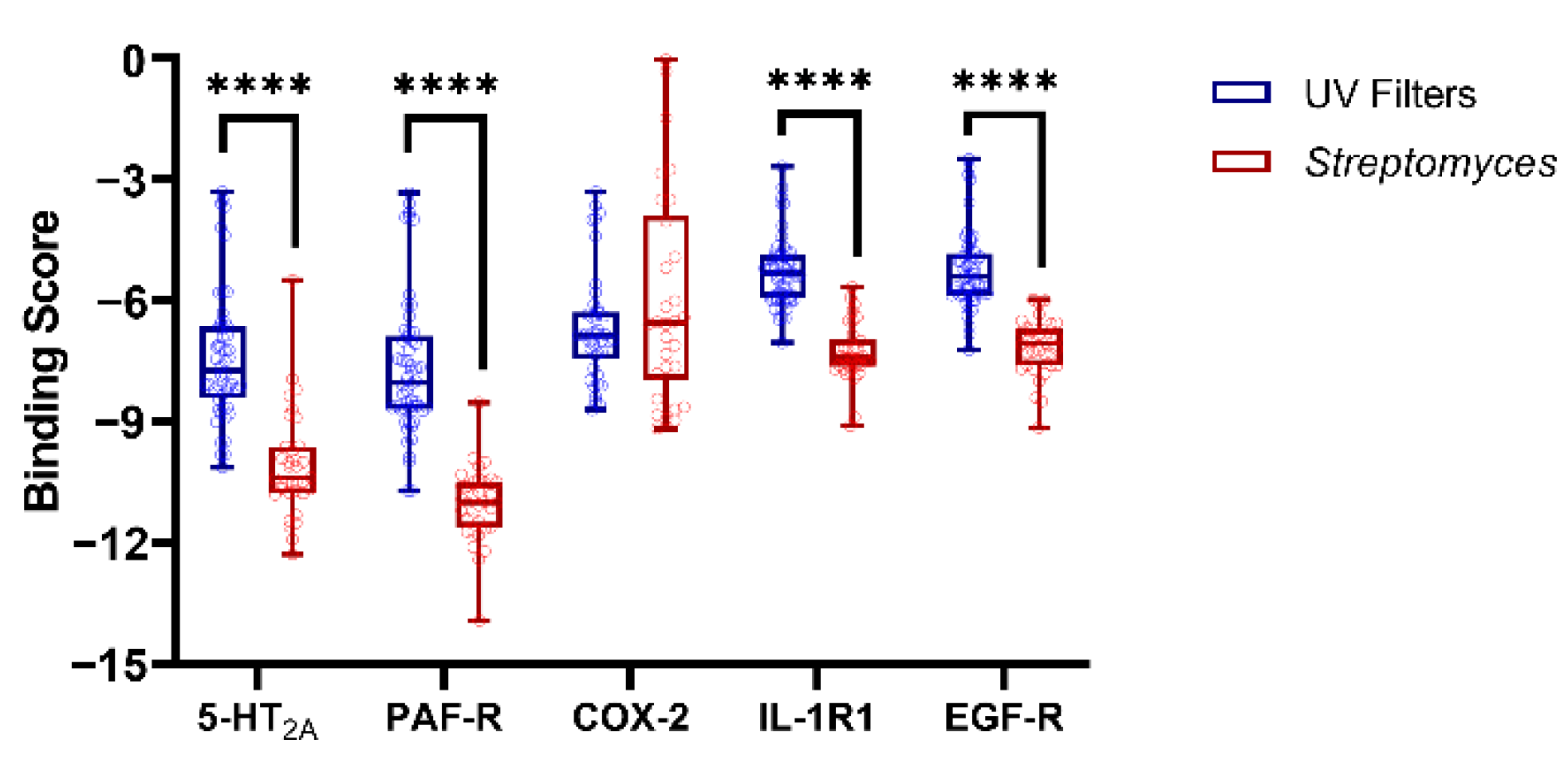
3.2. Molecular Docking Studies: Target-Based Virtual Screening
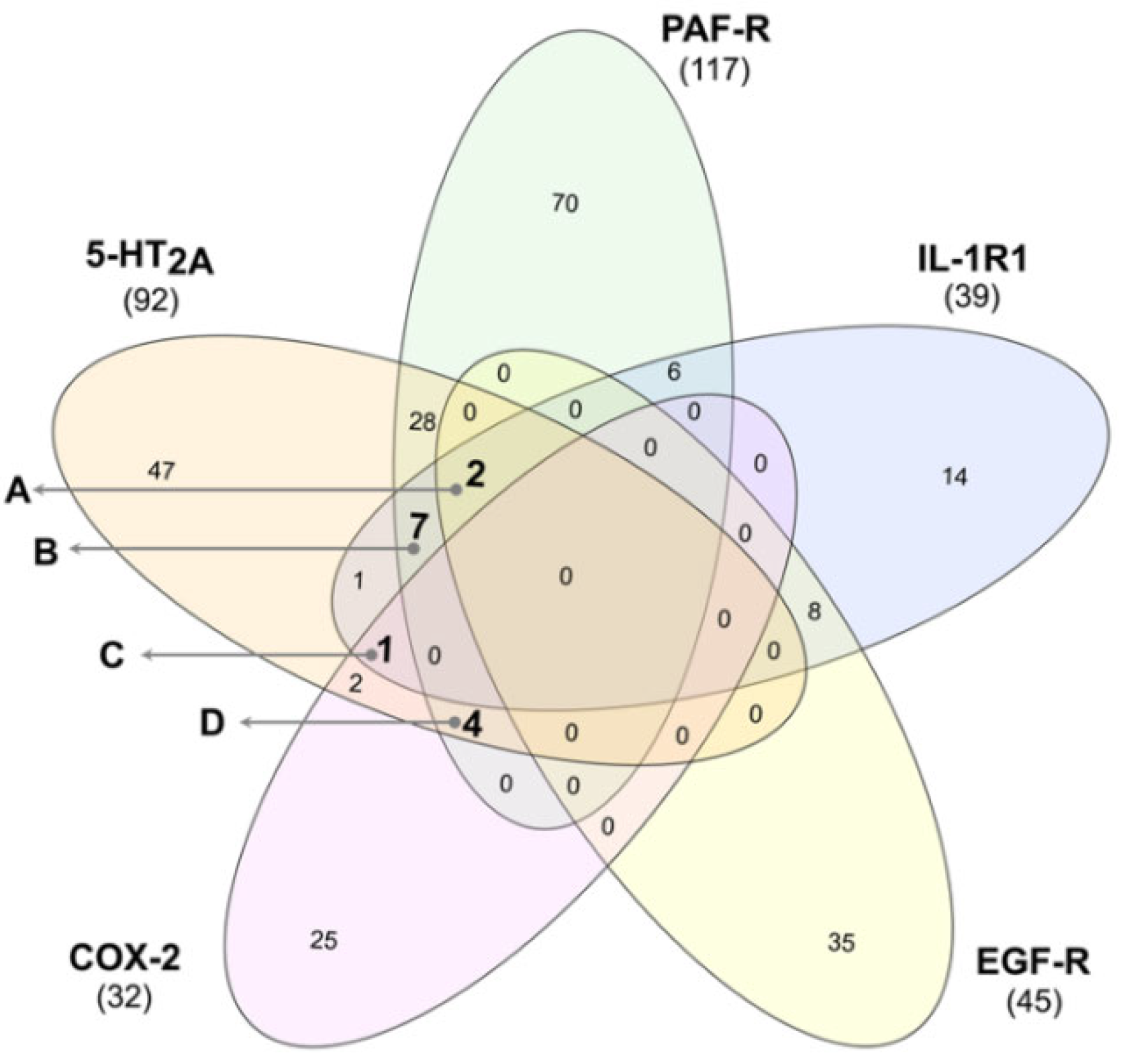
3.3. Identification of Multi-Target Ligands
- Intersect A: two compounds with high affinity for 5-HT2A, PAF-R, IL-1R1, and EGF-R.
- Intersect B: seven compounds with high affinity for 5-HT2A, PAF-R, and IL-1R1.
- Intersect C: a compound with high affinity for 5-HT2A, IL-1R1, and COX-2.
- Intersect D: four compounds with high affinity for 5-HT2A, PAF-R, and COX-2.
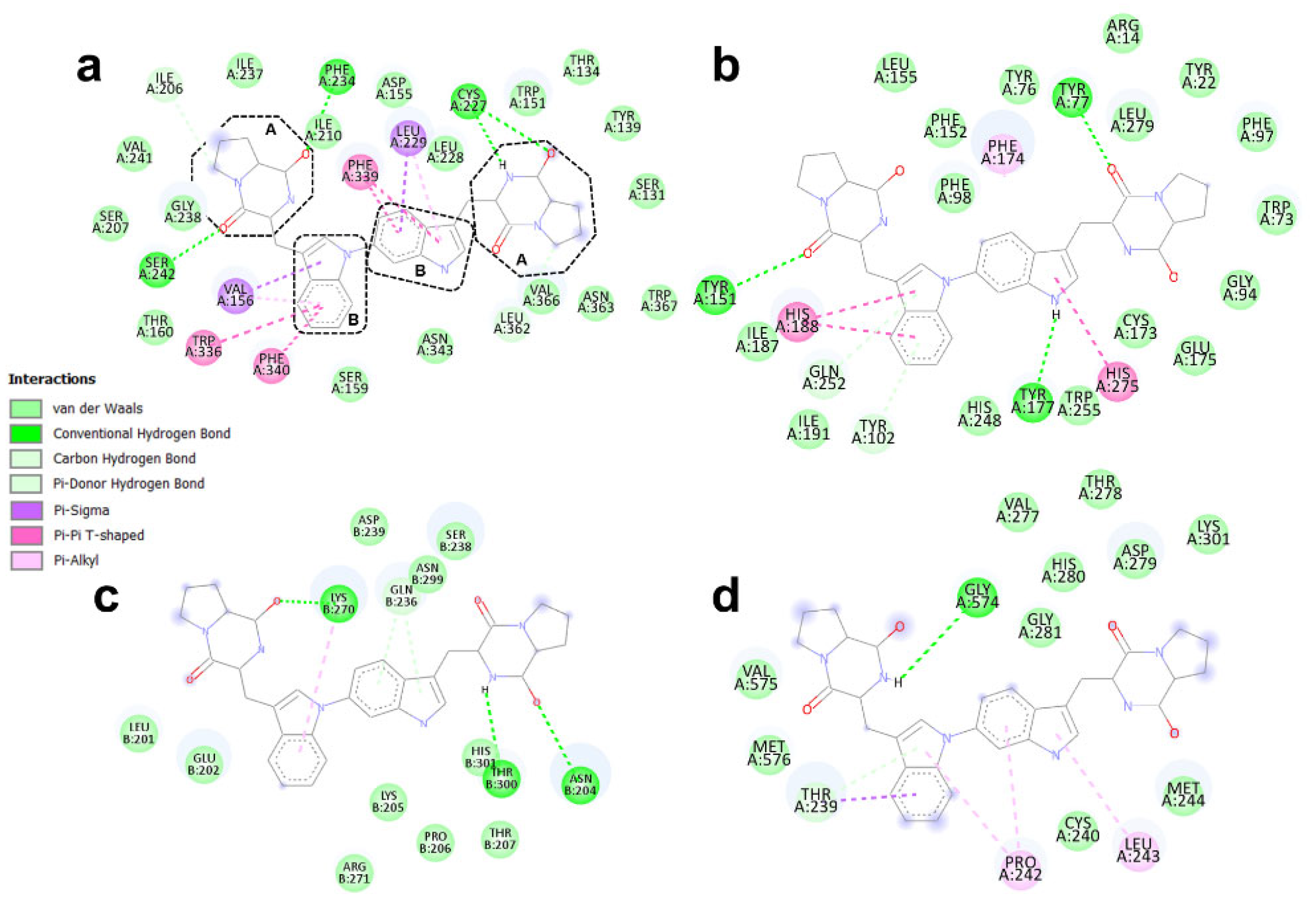
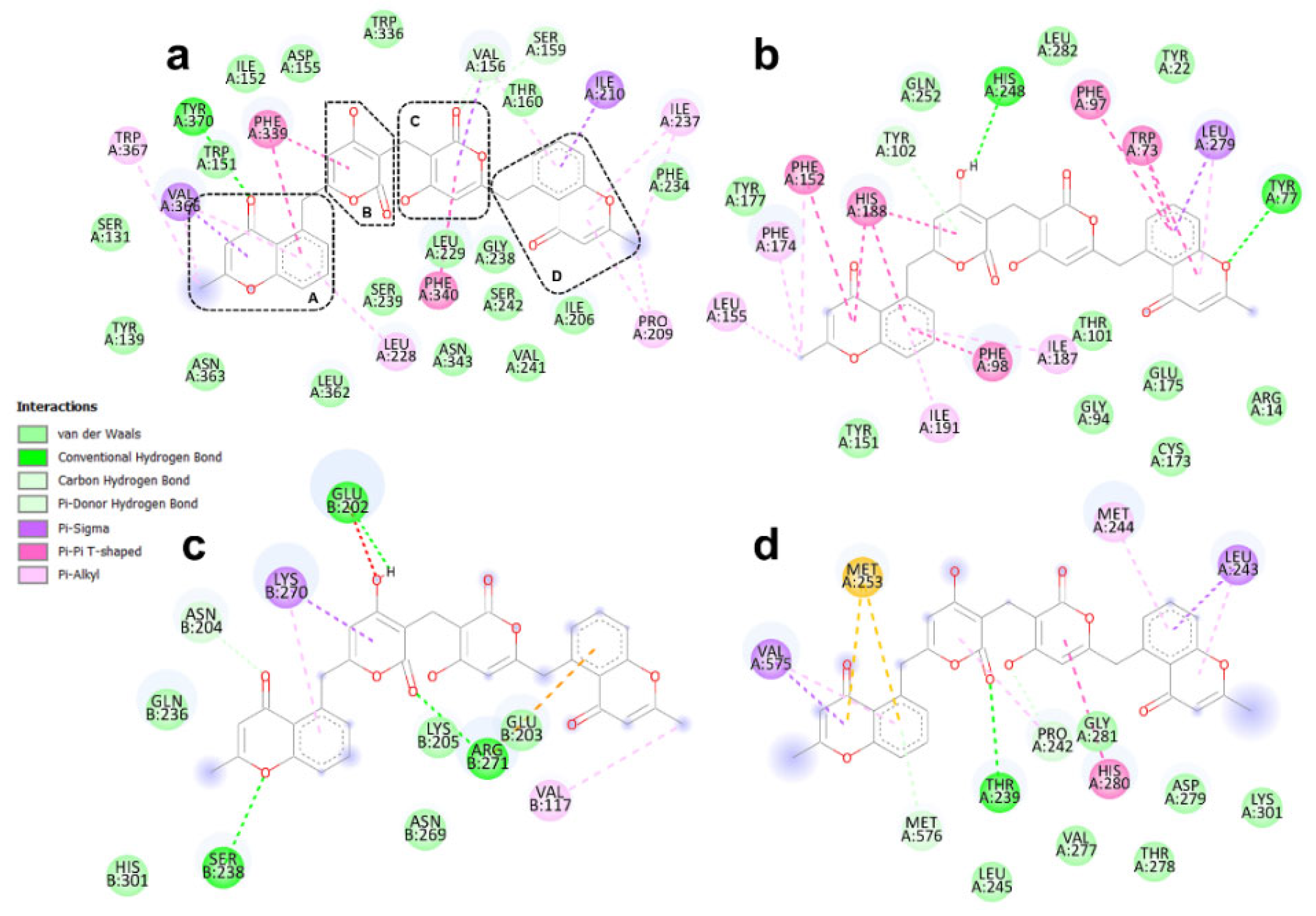
3.4. Binding Modes of Aspergilazine A and Phaeochromycin F
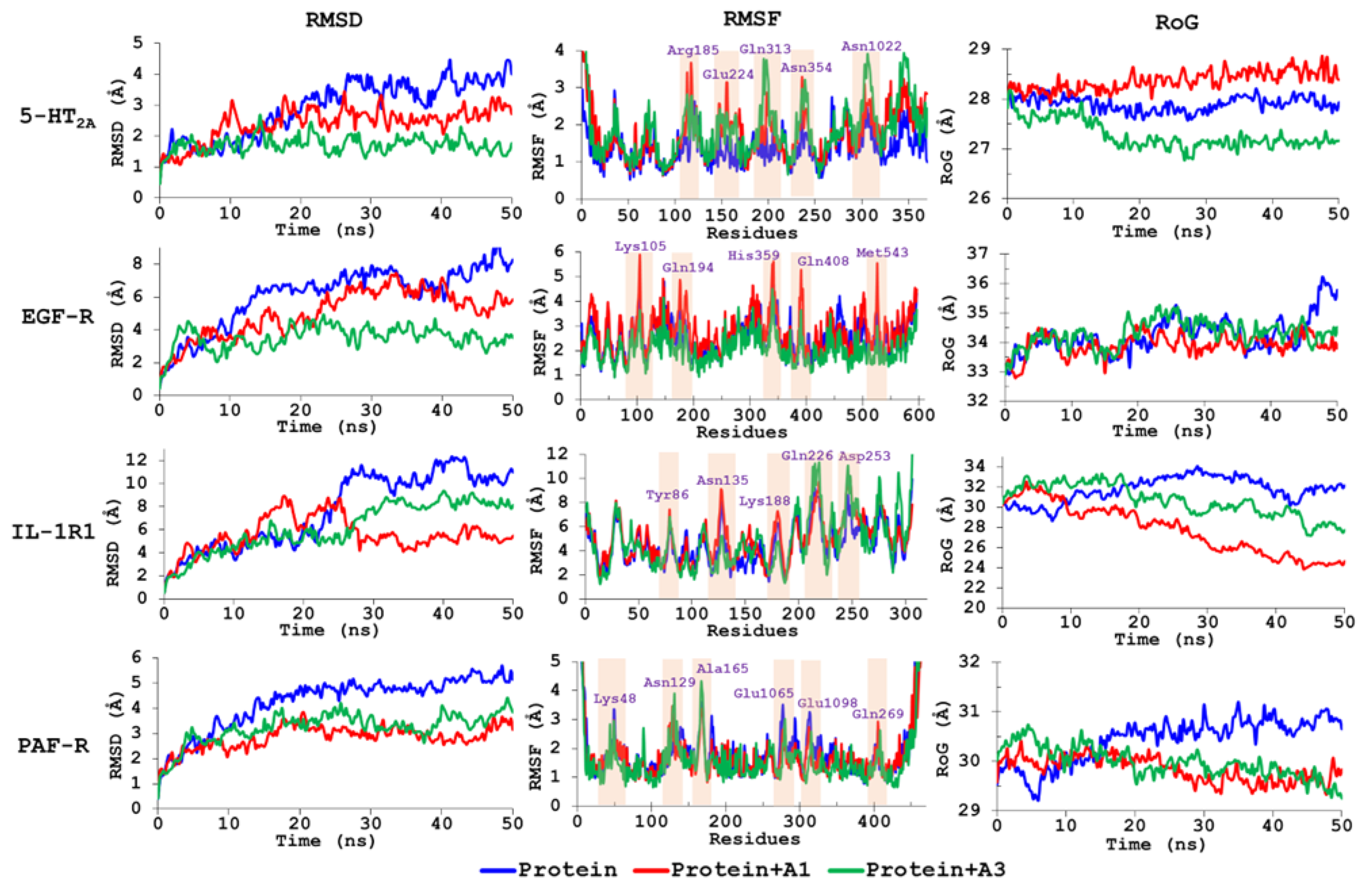
3.5. Molecular Dynamics Simulations
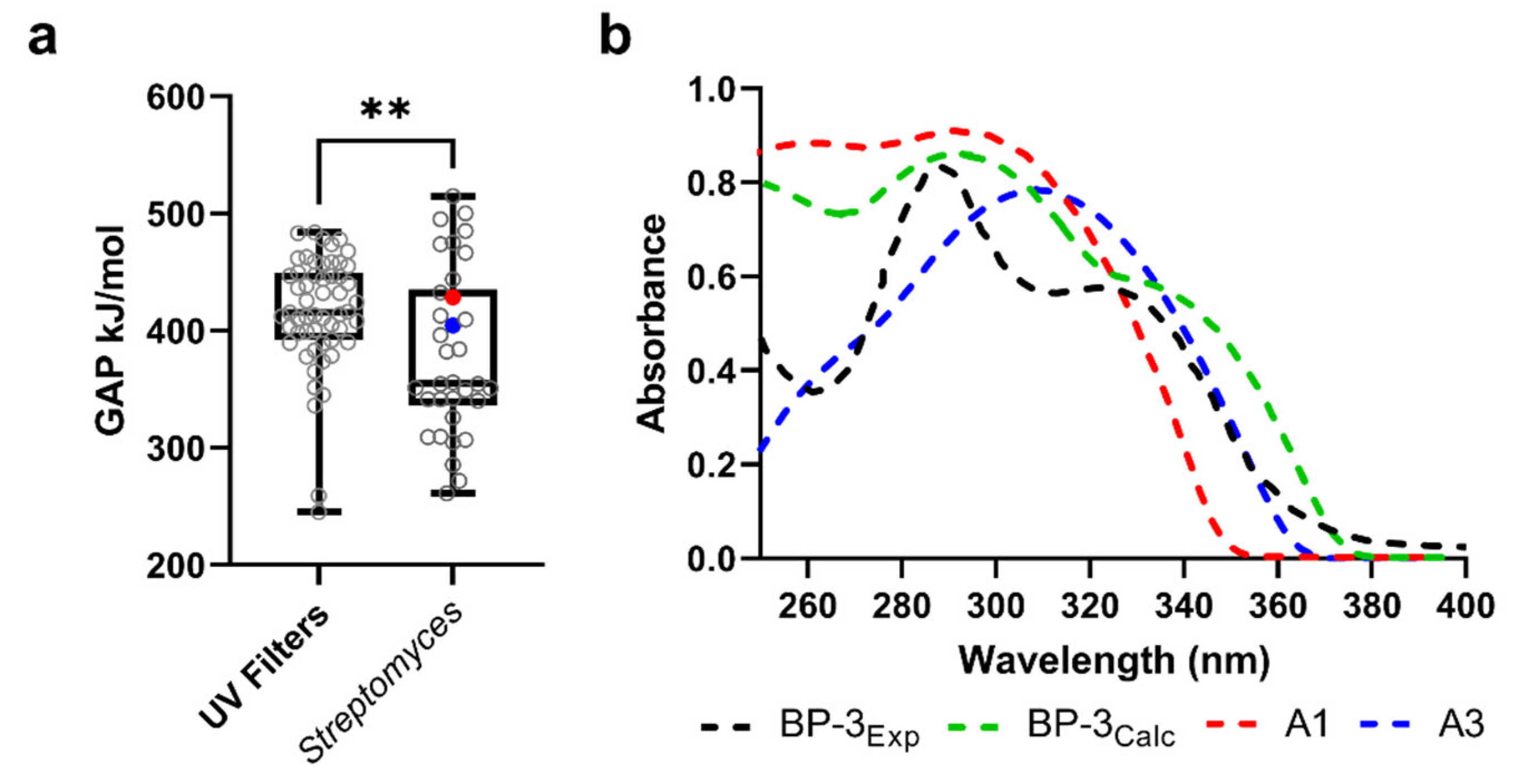
3.6. UV-Absorbing Profile of the Selected Multi-Target Compounds
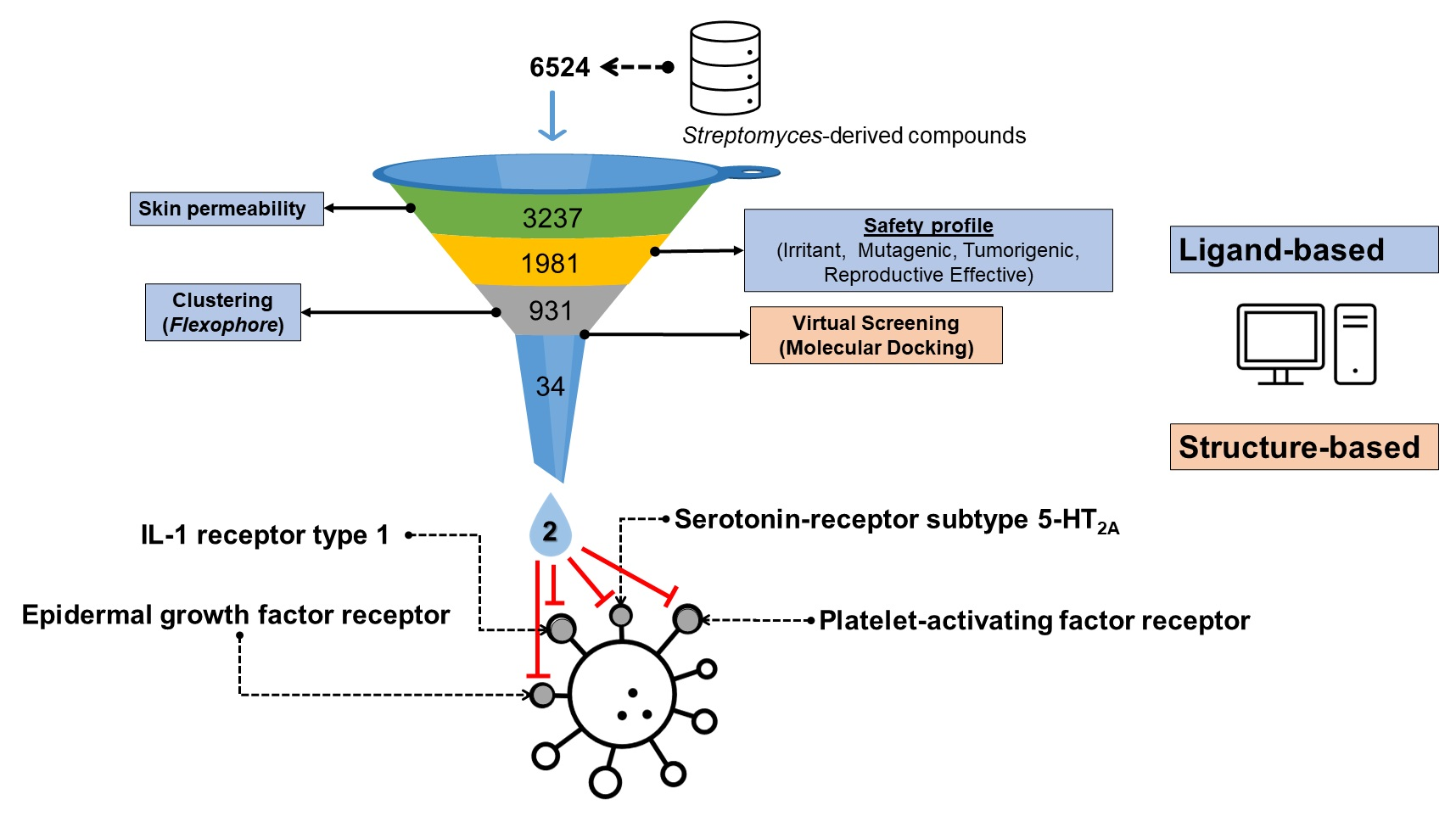
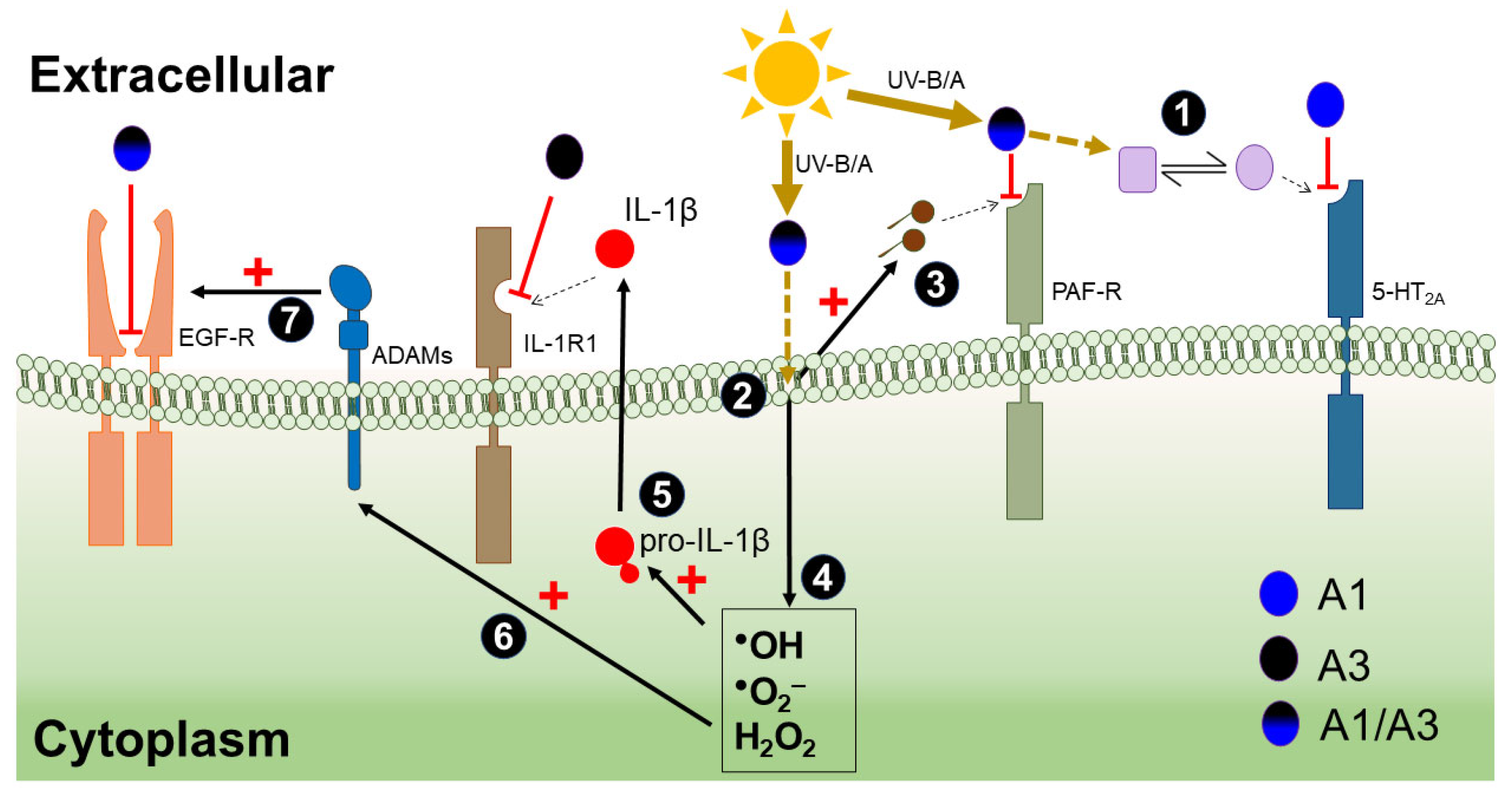
4. Discussion
- (i.)
- Mitigating the amount of radiation that would reach the cells by absorption.
- (ii.)
- (iii.)
- (iv.)
- (v.)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilchrest, B.A. Actinic Injury. Annu. Rev. Med. 1990, 41, 199–210. [Google Scholar] [CrossRef]
- Moan, J.; Grigalavicius, M.; Baturaite, Z.; Dahlback, A.; Juzeniene, A. The relationship between UV exposure and incidence of skin cancer. Photodermatol. Photoimmunol. Photomed. 2015, 31, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.C.; Seebode, C.; Lehmann, J.; Emmert, S. Photocarcinogenesis and Skin Cancer Prevention Strategies: An Update. Anticancer Res. 2018, 38, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Arellano-Mendoza, M.I.; Stengel, F. Current challenges in photoprotection. J. Am. Acad. Dermatol. 2017, 76, S91–S99. [Google Scholar] [CrossRef] [PubMed]
- Paiva, J.P.; Diniz, R.R.; Leitão, A.C.; Cabral, L.M.; Fortunato, R.S.; Santos, B.A.M.C.; de Pádula, M. Insights and controversies on sunscreen safety. Crit. Rev. Toxicol. 2020, 50, 707–723. [Google Scholar] [CrossRef]
- Schaap, I.; Slijkerman, D.M.E. An environmental risk assessment of three organic UV-filters at Lac Bay, Bonaire, Southern Caribbean. Mar. Pollut. Bull. 2018, 135, 490–495. [Google Scholar] [CrossRef]
- Kaiser, D.; Sieratowicz, A.; Zielke, H.; Oetken, M.; Hollert, H.; Oehlmann, J. Ecotoxicological effect characterisation of widely used organic UV filters. Environ. Pollut. 2012, 163, 84–90. [Google Scholar] [CrossRef]
- Tsui, M.M.P.; Leung, H.W.; Wai, T.C.; Yamashita, N.; Taniyasu, S.; Liu, W.; Lam, P.K.S.; Murphy, M.B. Occurrence, distribution and ecological risk assessment of multiple classes of UV filters in surface waters from different countries. Water Res. 2014, 67, 55–65. [Google Scholar] [CrossRef]
- Tsui, M.M.P.; Lam, J.C.W.; Ng, T.Y.; Ang, P.O.; Murphy, M.B.; Lam, P.K.S. Occurrence, Distribution, and Fate of Organic UV Filters in Coral Communities. Environ. Sci. Technol. 2017, 51, 4182–4190. [Google Scholar] [CrossRef]
- Lozano, C.; Givens, J.; Stien, D.; Matallana-Surget, S.; Lebaron, P. Bioaccumulation and Toxicological Effects of UV-Filters on Marine Species. In Sunscreens in Coastal Ecosystems; Springer International Publishing: Cham, Switzerland, 2020; pp. 85–130. [Google Scholar]
- Poon, F.; Kang, S.; Chien, A.L. Mechanisms and treatments of photoaging. Photodermatol. Photoimmunol. Photomed. 2015, 31, 65–74. [Google Scholar] [CrossRef]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Nasti, T.H.; Timares, L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem. Photobiol. 2012, 88, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFκB and TNFα signal transduction pathways. Arch. Dermatol. Res. 2010, 302, 5–17. [Google Scholar] [CrossRef] [PubMed]
- El-Abaseri, T.B.; Hansen, L.A. EGFR Activation and Ultraviolet Light-Induced Skin Carcinogenesis. J. Biomed. Biotechnol. 2007, 2007, 1–4. [Google Scholar] [CrossRef]
- Rundhaug, J.E.; Mikulec, C.; Pavone, A.; Fischer, S.M. A role for cyclooxygenase-2 in ultraviolet light-induced skin carcinogenesis. Mol. Carcinog. 2007, 46, 692–698. [Google Scholar] [CrossRef]
- Pinnell, S.R. Cutaneous photodamage, oxidative stress, and topical antioxidant protection. J. Am. Acad. Dermatol. 2003, 48, 1–22. [Google Scholar] [CrossRef]
- Nichols, J.A.; Katiyar, S.K. Skin photoprotection by natural polyphenols: Anti-inflammatory, antioxidant and DNA repair mechanisms. Arch. Dermatol. Res. 2010, 302, 71–83. [Google Scholar] [CrossRef]
- Shaath, N.A. Ultraviolet filters. Photochem. Photobiol. Sci. 2010, 9, 464. [Google Scholar] [CrossRef]
- Shaath, N.A. The Chemistry of Ultraviolet Filters. In Principles and Practice of Photoprotection; Springer International Publishing: Cham, Switzerland, 2016; pp. 143–157. ISBN 9783319293820. [Google Scholar]
- Subhadarshani, S.; Athar, M.; Elmets, C.A. Photocarcinogenesis. Curr. Dermatol. Rep. 2020, 9, 189–199. [Google Scholar] [CrossRef]
- Giacomoni, P.U. Appropriate Technologies to Accompany Sunscreens in the Battle Against Ultraviolet, Superoxide, and Singlet Oxygen. Antioxidants 2020, 9, 1091. [Google Scholar] [CrossRef]
- Romanhole, R.C.; Fava, A.L.M.; Tundisi, L.L.; de Macedo, L.M.; dos Santos, É.M.; Ataide, J.A.; Mazzola, P.G. Unplanned absorption of sunscreen ingredients: Impact of formulation and evaluation methods. Int. J. Pharm. 2020, 591, 120013. [Google Scholar] [CrossRef]
- Oral, D.; Yirun, A.; Erkekoglu, P. Safety Concerns of Organic Ultraviolet Filters: Special Focus on Endocrine-Disrupting Properties. J. Environ. Pathol. Toxicol. Oncol. 2020, 39, 201–212. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Lin, B.; Tao, Y. Whole-cell biocatalysts by design. Microb. Cell Fact. 2017, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Belknap, K.C.; Park, C.J.; Barth, B.M.; Andam, C.P. Genome mining of biosynthetic and chemotherapeutic gene clusters in Streptomyces bacteria. Sci. Rep. 2020, 10, 2003. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Suárez, J.; Coy-Barrera, E.; Villamil, L.; Díaz, L. Streptomyces-Derived Metabolites with Potential Photoprotective Properties—A Systematic Literature Review and Meta-Analysis on the Reported Chemodiversity. Molecules 2020, 25, 3221. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Brogi, S.; Ramalho, T.C.; Kuca, K.; Medina-Franco, J.L.; Valko, M. Editorial: In silico Methods for Drug Design and Discovery. Front. Chem. 2020, 8, 612. [Google Scholar] [CrossRef] [PubMed]
- Phatak, S.S.; Stephan, C.C.; Cavasotto, C.N. High-throughput and in silico screenings in drug discovery. Expert Opin. Drug Discov. 2009, 4, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Elokely, K.M.; Doerksen, R.J. Docking Challenge: Protein Sampling and Molecular Docking Performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef]
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef]
- Moumbock, A.F.A.; Gao, M.; Qaseem, A.; Li, J.; Kirchner, P.A.; Ndingkokhar, B.; Bekono, B.D.; Simoben, C.V.; Babiaka, S.B.; Malange, Y.I.; et al. StreptomeDB 3.0: An updated compendium of streptomycetes natural products. Nucleic Acids Res. 2021, 49, D600–D604. [Google Scholar] [CrossRef] [PubMed]
- Krämer, M.; Sachsenmaier, C.; Herrlich, P.; Rahmsdorf, H.J. UV irradiation-induced interleukin-1 and basic fibroblast growth factor synthesis and release mediate part of the UV response. J. Biol. Chem. 1993, 268, 6734–6741. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Yang, C.Y. Identification of potential small molecule allosteric modulator sites on IL-1R1 ectodomain using accelerated conformational sampling method. PLoS ONE 2015, 10, e0118671. [Google Scholar] [CrossRef]
- Yang, R.Y.C.; Yang, K.S.; Pike, L.J.; Marshall, G.R. Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chem. Biol. Drug Des. 2010, 76, 1–9. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Nohynek, G.J.; Schaefer, H. Benefit and risk of organic ultraviolet filters. Regul. Toxicol. Pharmacol. 2001, 33, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, B.M.; Pugh, W.J.; Roberts, M.S. Simple rules defining the potential of compounds for transdermal delivery or toxicity. Pharm. Res. 2004, 21, 1047–1054. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Couteau, C.; Chauvet, C.; Paparis, E.; Coiffard, L. UV Filters, Ingredients with a Recognized Anti-Inflammatory Effect. PLoS ONE 2012, 7, e46187. [Google Scholar] [CrossRef]
- Dougherty, D.A. The Cation−π Interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Petukhov, M.; Uegaki, K.; Yumoto, N.; Yoshikawa, S.; Serrano, L. Position dependence of amino acid intrinsic helical propensities II: Non-charged polar residues: Ser, Thr, Asn, and Gln. Protein Sci. 1999, 8, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Cochran, D.A.E.; Doig, A.J. Effect of the N2 residue on the stability of the α-helix for all 20 amino acids. Protein Sci. 2008, 10, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Raffa, R.B.; Pergolizzi, J.V.; Taylor, R.; Kitzen, J.M. Sunscreen bans: Coral reefs and skin cancer. J. Clin. Pharm. Ther. 2019, 44, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Springsteen, A.; Yurek, R.; Frazier, M.; Carr, K.F. In vitro measurement of sun protection factor of sunscreens by diffuse transmittance. Anal. Chim. Acta 1999, 380, 155–164. [Google Scholar] [CrossRef]
- Srivastava, P.; Tiwari, A. Critical Role of Computer Simulations in Drug Discovery and Development. Curr. Top. Med. Chem. 2017, 17, 2422–2432. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Korkina, L.G. Current Trends in Medicinal Chemistry of Photoprotection and Phototherapy. Curr. Med. Chem. 2018, 25, 5466–5468. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Wondrak, G.T. TLR4-directed Molecular Strategies Targeting Skin Photodamage and Carcinogenesis. Curr. Med. Chem. 2019, 25, 5487–5502. [Google Scholar] [CrossRef]
- Tadano, S.; Sugimachi, Y.; Sumimoto, M.; Tsukamoto, S.; Ishikawa, H. Collective Synthesis and Biological Evaluation of Tryptophan-Based Dimeric Diketopiperazine Alkaloids. Chem.-Eur. J. 2016, 22, 1277–1291. [Google Scholar] [CrossRef]
- Li, J.; Lu, C.-H.; Zhao, B.-B.; Zheng, Z.-H.; Shen, Y.-M. Phaeochromycins F–H, three new polyketide metabolites from Streptomyces sp. DSS-18. Beilstein J. Org. Chem. 2008, 4, 46. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Zhang, H.J.; Li, J.Q.; Ding, W.J.; Ma, Z.J. Bioactive Indolocarbazoles from the Marine-Derived Streptomyces sp. DT-A61. J. Nat. Prod. 2018, 81, 949–956. [Google Scholar] [CrossRef]
- Rab, E.; Kekos, D.; Roussis, V.; Ioannou, E. α-Pyrone Polyketides from Streptomyces ambofaciens BI0048, an Endophytic Actinobacterial Strain Isolated from the Red Alga Laurencia glandulifera. Mar. Drugs 2017, 15, 389. [Google Scholar] [CrossRef]
- Seebode, C.; Lehmann, J.; Emmert, S. Photocarcinogenesis and Skin Cancer Prevention Strategies. Anticancer Res. 2016, 36, 1371–1378. [Google Scholar] [PubMed]
- Liu, J.; Gu, B.; Yang, L.; Yang, F.; Lin, H. New Anti-inflammatory Cyclopeptides From a Sponge-Derived Fungus Aspergillus violaceofuscus. Front. Chem. 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Liu, X.; Zhang, Q.; Deng, Y.; Zang, Y.; Wang, J.; Liu, J.; Zhou, Q.; Hu, L.; Zhu, H.; et al. Three New Indole Diketopiperazine Alkaloids from Aspergillus ochraceus. Chem. Biodivers. 2018, 15, e1700550. [Google Scholar] [CrossRef] [PubMed]
- Bulger, E.M.; Arbabi, S.; Garcia, I.; Maier, R.V. The macrophage response to endotoxin requires platelet activating factor. Shock 2002, 17, 173–179. [Google Scholar] [CrossRef]
- Im, S.Y.; Han, S.J.; Ko, H.M.; Choi, J.H.; Chun, S.B.; Lee, D.G.; Ha, T.Y.; Lee, H.-K. Involvement of nuclear factor-xB in platelet-activating factor-mediated tumor necrosis factor-α expression. Eur. J. Immunol. 1997, 27, 2800–2804. [Google Scholar] [CrossRef]
- Liu, Q.; Mu, Y.; An, Q.; Xun, J.; Ma, J.; Wu, W.; Xu, M.; Xu, J.; Han, L.; Huang, X. Total synthesis and anti-inflammatory evaluation of violacin A and its analogues. Bioorg. Chem. 2020, 94, 103420. [Google Scholar] [CrossRef]
- Graziani, E.I.; Ritacco, F.V.; Bernan, V.S.; Telliez, J.-B. Phaeochromycins A−E, Anti-inflammatory Polyketides Isolated from the Soil Actinomycete Streptomyces p haeochromogenes LL-P018. J. Nat. Prod. 2005, 68, 1262–1265. [Google Scholar] [CrossRef]
- Kimura, K.T.; Asada, H.; Inoue, A.; Kadji, F.M.N.; Im, D.; Mori, C.; Arakawa, T.; Hirata, K.; Nomura, Y.; Nomura, N.; et al. Structures of the 5-HT 2A receptor in complex with the antipsychotics risperidone and zotepine. Nat. Struct. Mol. Biol. 2019, 26, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Nussinov, R.; Keskin, O. The Structural Pathway of Interleukin 1 (IL-1) Initiated Signaling Reveals Mechanisms of Oncogenic Mutations and SNPs in Inflammation and Cancer. PLoS Comput. Biol. 2014, 10, e1003470. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Walker, F.; Orchard, S.G.; Jorissen, R.N.; Hall, N.E.; Zhang, H.-H.; Hoyne, P.A.; Adams, T.E.; Johns, T.G.; Ward, C.; Garrett, T.P.J.; et al. CR1/CR2 Interactions Modulate the Functions of the Cell Surface Epidermal Growth Factor Receptor. J. Biol. Chem. 2004, 279, 22387–22398. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H. EGFR ligands and their signaling scissors, ADAMs, as new molecular targets for anticancer treatments. J. Dermatol. Sci. 2009, 56, 148–153. [Google Scholar] [CrossRef]
- Singh, B.; Schneider, M.; Knyazev, P.; Ullrich, A. UV-induced EGFR signal transactivation is dependent on proligand shedding by activated metalloproteases in skin cancer cell lines. Int. J. Cancer 2009, 124, 531–539. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
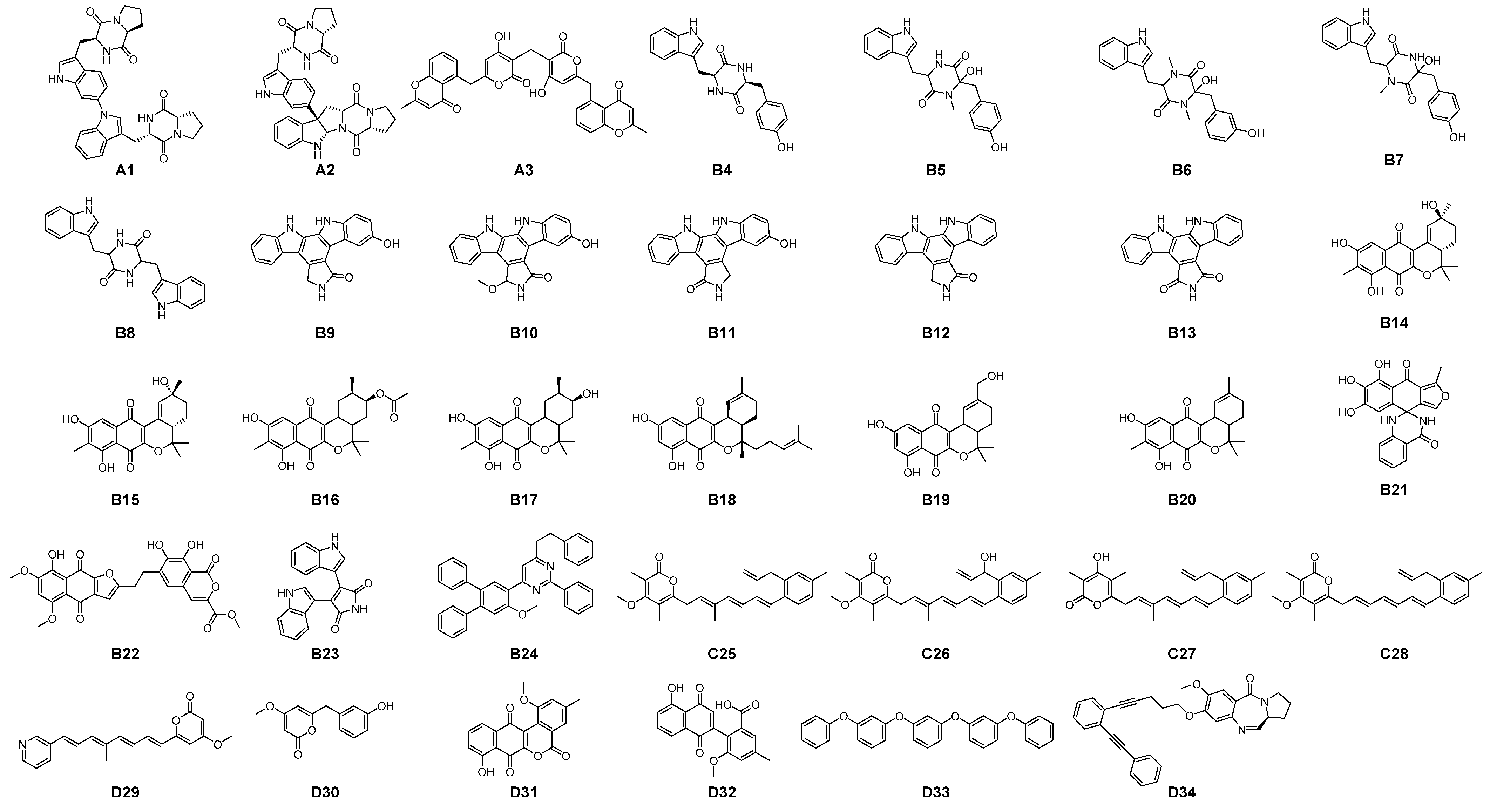{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | 5−HT2A BS a | RSD b | IL−1R1 BS a | RSD b | PAF−R BS a | RSD b | EGF−R BS a | RSD b |
|---|---|---|---|---|---|---|---|---|
| A1 | −11.49 ± 0.03 | 0.28 | −8.88 ± 0.28 | 3.13 | −12.20 ± 0.00 | 0.00 | −8.50 ± 0.00 | 0.00 |
| A2 | −5.50 ± 0.00 | 0.00 | −9.10 ± 0.00 | 0.00 | −12.40 ± 0.05 | 0.38 | −9.14 ± 0.13 | 1.38 |
| A3 | −11.30 ± 0.00 | 0.00 | −7.91 ± 0.03 | 0.40 | −11.46 ± 0.05 | 0.45 | −8.40 ± 0.00 | 0.00 |
| Ligand | 5−HT2A BS a | RSD b | IL−1R1 BS a | RSD b | PAF−R BS a | RSD b |
|---|---|---|---|---|---|---|
| B4 | −10.00 ± 0.00 | 0.00 | −7.50 ± 0.00 | 0.00 | −11.00 ± 0.00 | 0.00 |
| B5 | −9.90 ± 0.00 | 0.00 | −7.81 ± 0.03 | 0.40 | −10.30 ± 0.00 | 0.00 |
| B6 | −9.60 ± 0.00 | 0.00 | −7.40 ± 0.00 | 0.00 | −11.06 ± 0.05 | 0.47 |
| B7 | −9.61 ± 0.03 | 0.33 | −7.00 ± 0.00 | 0.00 | −10.50 ± 0.32 | 3.01 |
| B8 | −10.71 ± 0.03 | 0.30 | −7.74 ± 0.21 | 2.67 | −11.80 ± 0.00 | 0.00 |
| B9 | −10.51 ± 0.03 | 0.30 | −7.60 ± 0.00 | 0.00 | −11.70 ± 0.00 | 0.00 |
| B10 | −10.40 ± 0.00 | 0.00 | −7.69 ± 0.03 | 0.41 | −11.39 ± 0.03 | 0.28 |
| B11 | −10.80 ± 0.00 | 0.00 | −7.51 ± 0.03 | 0.42 | −11.41 ± 0.03 | 0.28 |
| B12 | −10.66 ± 0.05 | 0.48 | −7.60 ± 0.00 | 0.00 | −11.60 ± 0.00 | 0.00 |
| B13 | −10.53 ± 0.05 | 0.46 | −7.80 ± 0.00 | 0.00 | −12.10 ± 0.00 | 0.00 |
| B14 | −10.37 ± 0.41 | 3.96 | −7.50 ± 0.00 | 0.00 | −10.80 ± 0.00 | 0.00 |
| B15 | −8.71 ± 0.03 | 0.36 | −7.18 ± 0.38 | 5.29 | −10.40 ± 0.00 | 0.00 |
| B16 | −10.00 ± 0.00 | 0.00 | −7.19 ± 0.37 | 5.10 | −10.58 ± 0.04 | 0.40 |
| B17 | −9.70 ± 0.00 | 0.00 | −7.23 ± 0.35 | 4.79 | −10.10 ± 0.00 | 0.00 |
| B18 | −7.94 ± 0.18 | 2.24 | −6.50 ± 0.17 | 2.61 | −11.19 ± 0.03 | 0.28 |
| B19 | −8.36 ± 0.05 | 0.62 | −6.85 ± 0.13 | 1.85 | −10.00 ± 0.00 | 0.00 |
| B20 | −8.89 ± 0.03 | 0.36 | −6.40 ± 0.00 | 0.00 | −10.90 ± 0.00 | 0.00 |
| B21 | −10.50 ± 0.00 | 0.00 | −7.59 ± 0.23 | 3.07 | −11.00 ± 0.00 | 0.00 |
| B22 | −10.50 ± 0.00 | 0.00 | −7.59 ± 0.06 | 0.75 | −10.97 ± 0.05 | 0.44 |
| B23 | −10.10 ± 0.00 | 0.00 | −7.69 ± 0.03 | 0.41 | −10.80 ± 0.00 | 0.00 |
| B24 | −11.91 ± 0.03 | 0.27 | −7.38 ± 0.04 | 0.57 | −13.91 ± 0.03 | 0.23 |
| Ligand | 5−HT2A BS a | RSD b | IL−1R1 BS a | RSD b | COX−2 BS a | RSD b |
|---|---|---|---|---|---|---|
| C25 | −10.76 ± 0.07 | 0.65 | −7.38 ± 0.04 | 0.57 | −8.93 ± 0.05 | 0.54 |
| C26 | −10.77 ± 0.05 | 0.45 | −7.42 ± 0.06 | 0.85 | −8.63 ± 0.05 | 0.56 |
| C27 | −11.45 ± 0.05 | 0.46 | −7.40 ± 0.00 | 0.00 | −8.83 ± 0.05 | 0.55 |
| C28 | −10.64 ± 0.07 | 0.66 | −7.19 ± 0.06 | 0.79 | −8.43 ± 0.15 | 1.77 |
| Ligand | 5−HT2A BS a | RSD b | PAF−R BS a | RSD b | COX−2 BS a | RSD b |
|---|---|---|---|---|---|---|
| D29 | −9.96 ± 0.05 | 0.52 | −10.59 ± 0.03 | 0.30 | −8.69 ± 0.03 | 0.36 |
| D30 | −8.20 ± 0.00 | 0.00 | −8.52 ± 0.04 | 0.49 | −7.80 ± 0.28 | 3.58 |
| D31 | −10.10 ± 0.00 | 0.00 | −10.75 ± 0.05 | 0.49 | −6.89 ± 1.80 | 26.06 |
| D32 | −8.81 ± 0.06 | 0.64 | −9.90 ± 0.00 | 0.00 | −6.01 ± 1.45 | 24.12 |
| D33 | −12.27 ± 0.05 | 0.39 | −11.71 ± 0.03 | 0.27 | −8.96 ± 0.05 | 0.58 |
| D34 | −11.59 ± 0.03 | 0.27 | −11.64 ± 0.07 | 0.60 | −9.16 ± 0.38 | 4.16 |
| Ligands | Targets | ΔEvdW a | ΔEele b | ΔGsol c | ΔGsasa d | ΔGbind e |
|---|---|---|---|---|---|---|
| A1 | 5−HT2A | −164.5 ± 1.8 | −139.4 ± 1.9 | 207.8 ± 1.5 | −22.3 ± 2.2 | −118.4 ± 1.9 |
| EGF−R | −141.8 ± 2.6 | −128.6 ± 1.7 | 187.6 ± 2.6 | −19.6 ± 2.3 | −102.4 ± 2.3 | |
| IL−1R1 | −207.7 ± 2.1 | −121.4 ± 2.2 | 144.3 ± 1.2 | −21.3 ± 2.4 | −206.1 ± 1.7 | |
| PAF−R | −216.9 ± 3.2 | −138.6 ± 2.4 | 208.6 ± 3.5 | −20.9 ± 2.1 | −167.8 ± 2.8 | |
| A3 | 5−HT2A | −211.8 ± 3.5 | −105.3 ± 1.9 | 180.3 ± 2.8 | −23.6 ± 2.0 | −160.4 ± 2.5 |
| EGF−R | −215.8 ± 3.4 | −112.5 ± 2.6 | 194.3 ± 2.4 | −24.1 ± 1.8 | −158.1 ± 2.7 | |
| IL−1R1 | −198.6 ± 2.8 | −109.7 ± 2.5 | 186.5 ± 2.9 | −23.9 ± 1.6 | −145.7 ± 2.4 | |
| PAF−R | −216.4 ± 1.9 | −111.2 ± 0.8 | 185.1 ± 3.1 | −21.4 ± 1.1 | −163.9 ± 1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Suárez, J.; Villamil, L.; Díaz, L.; Coy-Barrera, E. Uncovering Streptomyces-Derived Compounds as Cosmeceuticals for the Development of Improved Skin Photoprotection Products: An In Silico Approach to Explore Multi-Targeted Agents. Sci. Pharm. 2022, 90, 48. https://doi.org/10.3390/scipharm90030048
Sánchez-Suárez J, Villamil L, Díaz L, Coy-Barrera E. Uncovering Streptomyces-Derived Compounds as Cosmeceuticals for the Development of Improved Skin Photoprotection Products: An In Silico Approach to Explore Multi-Targeted Agents. Scientia Pharmaceutica. 2022; 90(3):48. https://doi.org/10.3390/scipharm90030048
Chicago/Turabian StyleSánchez-Suárez, Jeysson, Luisa Villamil, Luis Díaz, and Ericsson Coy-Barrera. 2022. "Uncovering Streptomyces-Derived Compounds as Cosmeceuticals for the Development of Improved Skin Photoprotection Products: An In Silico Approach to Explore Multi-Targeted Agents" Scientia Pharmaceutica 90, no. 3: 48. https://doi.org/10.3390/scipharm90030048
APA StyleSánchez-Suárez, J., Villamil, L., Díaz, L., & Coy-Barrera, E. (2022). Uncovering Streptomyces-Derived Compounds as Cosmeceuticals for the Development of Improved Skin Photoprotection Products: An In Silico Approach to Explore Multi-Targeted Agents. Scientia Pharmaceutica, 90(3), 48. https://doi.org/10.3390/scipharm90030048