
Phytochemical Standardization of an Extract Rich in Flavonoids from Flowers of Kalanchoe pinnata (Lam) Pers



Abstract
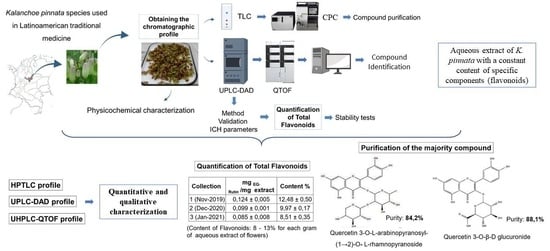
:
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Physicochemical Characterization of Plant Material
2.3. Chemical Composition of the Aqueous Extract of K. pinnata Flowers
2.4. Separation by Centrifugal Partition Chromatography (CPC)
2.5. Quantification of Total Flavonoids
2.5.1. Quantification by Colorimetric Method
2.5.2. Quantification by UPLC-PDA
2.6. Extract Stability under Stress Conditions
3. Results and Discussion
3.1. Physicochemical Characterization of Dried Flowers of K. pinnata
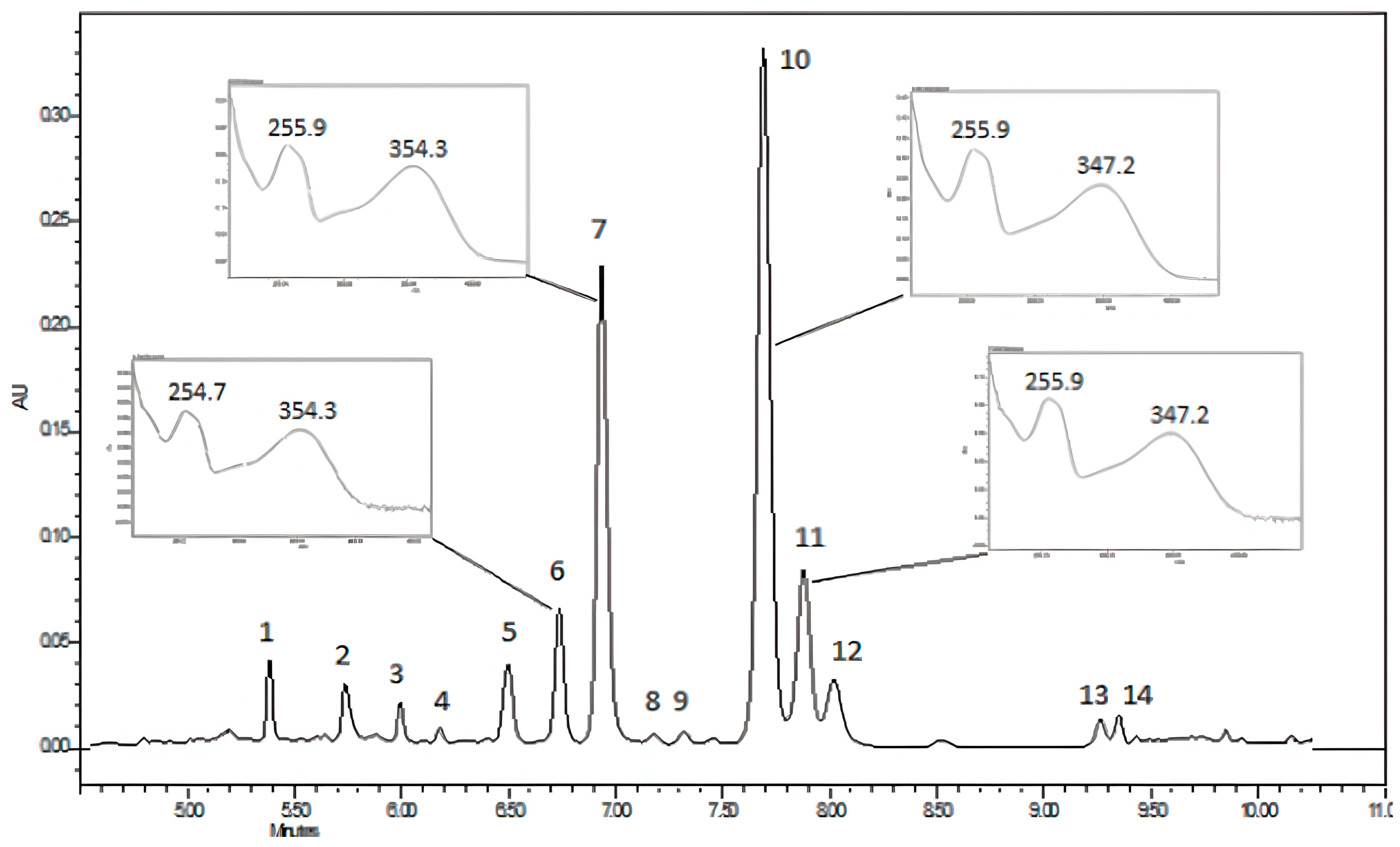
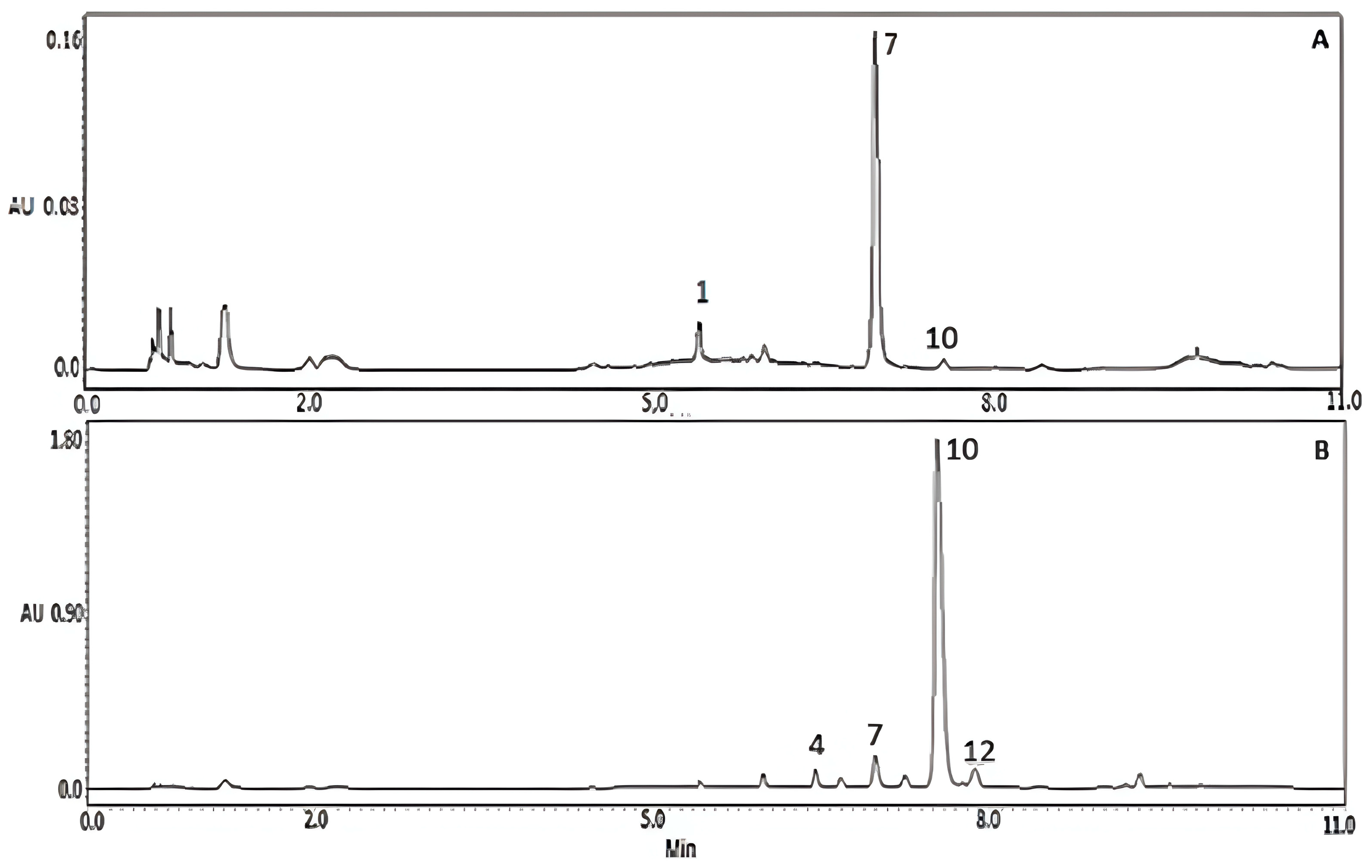
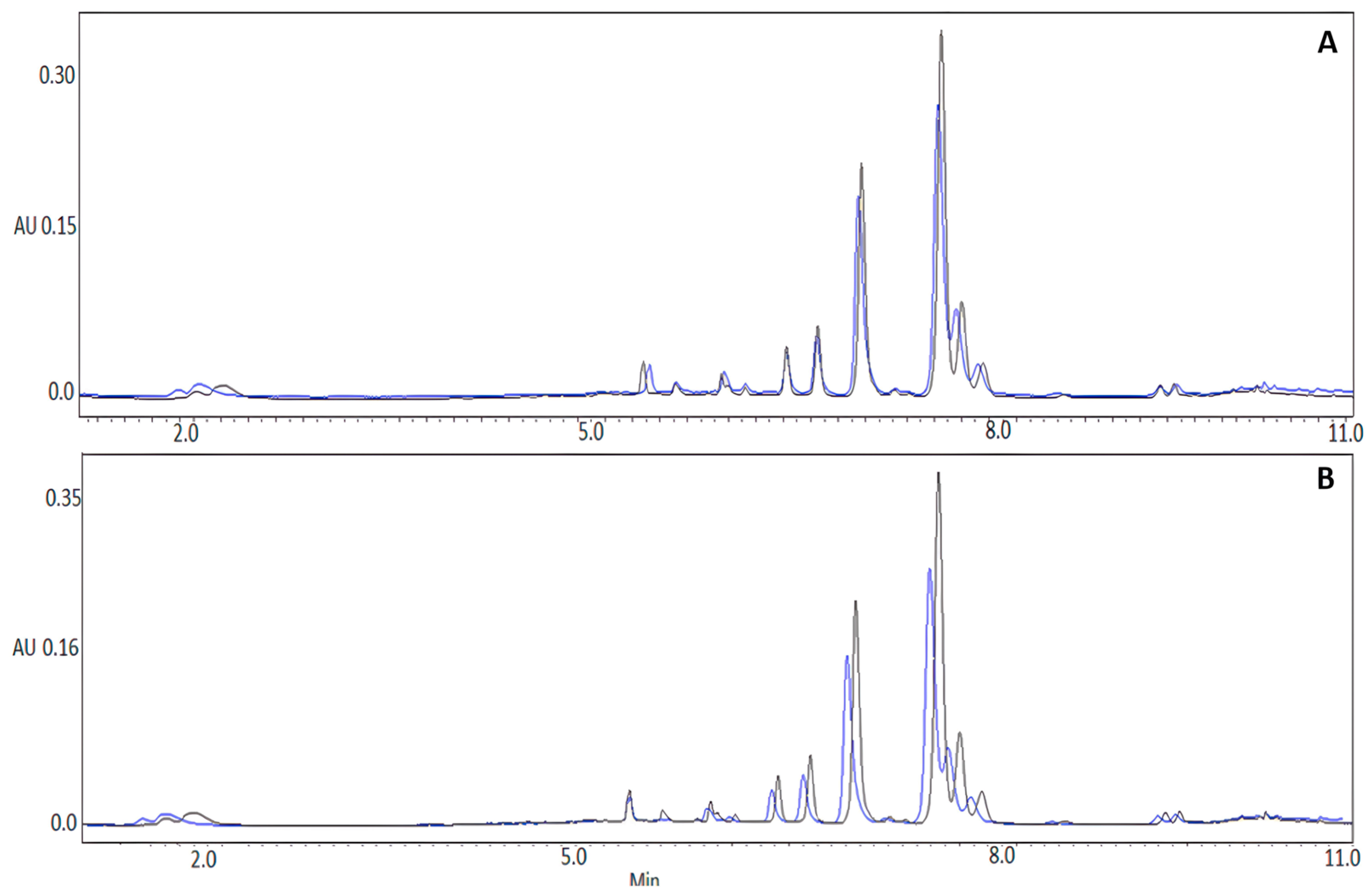
3.2. Chemical Composition of the Aqueous Extract of K. pinnata Flowers
3.3. Purification of Major Compounds by Centrifugal Partition Chromatography (CPC)
3.4. Quantification of Total Flavonoids
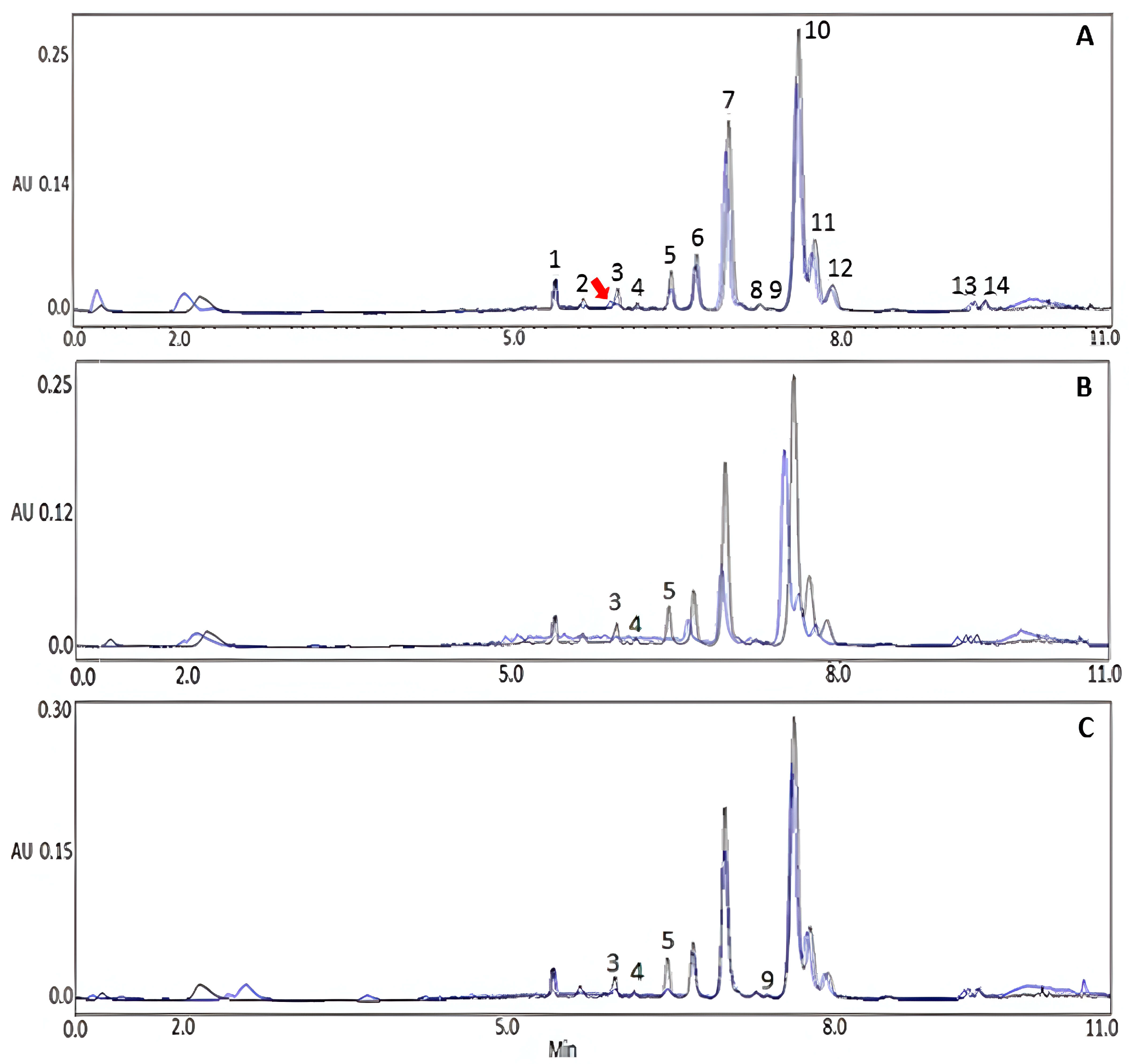
3.5. Extract Stability under Stress Conditions
3.5.1. Stability under Stress by Neutral Hydrolysis
3.5.2. Stability under Stress by Basic Hydrolysis (NaOH)
3.5.3. Stability under Stress by Acid Hydrolysis (HCl)
3.5.4. Extract Stability under Refrigeration Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polindara, Y.W.; Sanabria, O.L. Plantas y prácticas de conservación de la medicina tradicional en el suroriente de El Tambo, Cauca, Colombia. Bot. Sci. 2022, 100, 935–959. [Google Scholar] [CrossRef]
- Trujillo, W.; González, V. Plantas medicinales utilizadas por tres comunidades indígenas en el noroccidente de la Amazonia colombiana. Mundo Amaz. 2011, 2, 283–305. [Google Scholar]
- Nascimento, L.B.; de Aguiar, P.F.; Leal-Costa, M.V.; Coutinho, M.A.; Borsodi, M.P.G.; Rossi-Bergmann, B.; Costa, S. Optimization of Aqueous Extraction from Kalanchoe pinnata Leaves to Obtain the Highest Content of an Anti-inflammatory Flavonoid using a Response Surface Model. Phytochem. Anal. 2018, 29, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Harlalka, G.V.; Patil, C.R.; Patil, M.R. Protective effect of Kalanchoe pinnata pers. (Crassulaceae) on Gentamycin-induced nephrotoxicity in rats. Indian J. Pharmacol. 2007, 39, 201–205. [Google Scholar] [CrossRef]
- Ghasi, S.; Egwuib, C.; Achukwu, P.U.; Onyeanusi, J.C. Assessment of the medical benefit in the folkloric use of Bryophyllum Pinnatum leaf among the Igbos of Nigeria for the treatment of hypertension. Afr. J. Pharm. Pharmacol. 2011, 5, 83–92. [Google Scholar] [CrossRef]
- Okwu, D.E.; Nnamdi, F.U. Two novel flavonoids from Bryophyllum pinnatum and their antimicrobial Activity. J. Chem. Pharm. Res. 2011, 3, 1–10. [Google Scholar]
- Muzitano, F.; Bergonzi, M.; De Melo, G.; Lage, C.; Bilia, A.; Vincieri, F.; Rossi-Bergmann, B.; Costa, S. Influence of cultivation conditions, season of collection and extraction method on the content of antileishmanial flavonoids from Kalanchoe pinnata. J. Ethnopharmacol. 2011, 133, 132–137. [Google Scholar] [CrossRef]
- Nayak, B.S.; Marshall, J.R.; Isitor, G. Wound healing potential of ethanolic extract of Kalanchoe pinnata Lam. Leaf-A preliminary study. Indian J. Exp. Biol. 2010, 48, 572–576. [Google Scholar]
- Majaz, Q.A.; Nazim, S.; Asir, Q.; Shoeb, Q.; Bilal, G.M. Screening of In vitro anthelmentic activity of Kalanchoe pinnata roots. Int. J. Res. Ayurveda Pharm. 2011, 2, 221–223. [Google Scholar]
- Nguelefack, T.B.; Nana, P.; Atsamo, A.D.; Dimo, T.; Watcho, P.; Dongmo, A.B.; Tapondjou, L.A.; Njamen, D.; Wansi, S.L.; Kamanyi, A. Analgesic and anticonvulsant effects of extracts from the leaves of Kalanchoe crenata (Andrews) Haworth (Crassulaceae). J. Ethnopharmacol. 2006, 106, 70–75. [Google Scholar] [CrossRef]
- Mahata, S.; Maru, S.; Shukla, S.; Pandey, A.; Mugesh, G.; Das, B.; Bhart, A.C. Anticancer property of Bryophyllum pinnata (Lam.) Oken. leaf on human cervical cancer cells. Complement. Altern. Med. 2012, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Majaz, Q.A.; Tatiya, A.U.; Khurshid, M.; Nazim, S.; Siraj, S. The miracle plant (Kalanchoe pinnata): A phytochemical and pharmacological review. Int. J. Res. Ayurveda Pharm. 2011, 2, 1478–1482. [Google Scholar]
- Supratman, U.; Fujita, T.; Akiyama, K.; Hayashi, H.; Murakami, A.; Sakai, H.; Koshimizu, K.; Ohigashi, H. Anti-tumor promoting activity of bufadienolides from Kalanchoe pinnata and K. daigremontiana × tubiflora. Biosci. Biotechnol. Biochem. 2001, 65, 947–949. [Google Scholar] [CrossRef]
- Coutinho, M.A.; Muzitano, M.F.; Cruz, E.A.; Bergonzi, M.C.; Kaiser, C.R.; Tinoco, L.W.; Bilia, A.; Vincieri, F.; Bergmann, R.; Costa, S.S. Flowers from Kalanchoe pinnata are a Rich Source of T Cell-Suppressive Flavonoids. Nat. Prod. Commun. 2012, 7, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, L.B.; Leal-Costa, M.B.; Menezes, E.; Rodrigues, V.; Muzitano, M.; Costa, S.; Schwartz, E. Ultraviolet-B radiation effects on phenolic profile and flavonoid content of Kalanchoe pinnata. J. Photochem. Photobiol. B 2015, 148, 73–81. [Google Scholar] [CrossRef]
- Bogucka-Kocka, A.; Zidorn, C.; Kasprzycka, M.; Szymczak, G.; Szewczyk, K. Phenolic acid content, antioxidant and cytotoxic activities of four Kalanchoe species. Saudi J. Biol. Sci. 2018, 25, 622–630. [Google Scholar] [CrossRef]
- Kumar, S.B.; Chowdhury, A.; Das, J.S.; Hosen, S.M.; Uddin, R.; Rahaman, S. Literature review on pharmacological potentials of Kalanchoe pinnata (Crassulaceae). Afr. J. Pharm. Pharmacol. 2011, 5, 1258–1262. [Google Scholar] [CrossRef]
- Pereira, K.M.; Grecco, S.S.; Figueiredo, C.R.; Hosomi, J.K.; Nakamura, M.U.; João Henrique, G.L. Chemical composition and cytotoxicity of Kalanchoe pinnata leaves extracts prepared using Accelerated System Extraction (ASE). Nat. Prod. Commun. 2018, 13, 163–166. [Google Scholar] [CrossRef]
- Anupam, K.; Garima, V.; Roopak, K. Need of standardization of herbal medicines in modern era. Int. J. Phytomedicine 2016, 8, 300–330. [Google Scholar]
- Bijauliya, R.K.; Alok, S.; Chanchal, D.K.; Kumar, M. A comprehensive review on standardization of herbal drugs. Int. J. Pharm. Sci. Res. 2017, 8, 3663–3677. [Google Scholar]
- Echeverry, S. Aporte a la Estandarización del Proceso de Obtención de un Extracto de Hojas de Passiflora quadrangularis. Master’s Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2017. [Google Scholar]
- European Medicines Agency (EMEA). Guideline on Specifications: Test Procedures and Acceptance Criteria for Herbal Substances, Herbal Preparations, and Herbal Medicinal Products/Traditional Herbal Medicinal Products, EMEA/CVMP/815/00 Rev 1; European Medicines Agency: London, UK, 2006. [Google Scholar]
- Kunle, O.F.; Egharevba, M.O.; Ahmadu, P.O. Standardization of herbal medicines—A review. Int. J. Biodivers. Conserv. 2012, 4, 101–112. [Google Scholar] [CrossRef]
- Ochoa, A.; Marin, J.; Rivero, D.; Aguilera, E. Caracterización física, físico-química y química de extractos totales de hojas frescas de Petiveria alliacea L. con acción antimicrobiana. Rev. Mex. Cienc. Farm. 2013, 44, 52–59. [Google Scholar]
- Sharapin, N. Fundamentos de Tecnología de Productos Fitoterapéuticos; Pinzón, R., Ed.; CYTED: Santa fe de Bogotá, Colombia, 2000. [Google Scholar]
- World Health Organization (WHO). General Guidelines for Methodologies on Research and Evaluation of Traditional Medicine; WHO: Geneva, Switzerland, 2000. [Google Scholar]
- Wichtl, M. Herbal Drugs Phytopharmaceuticals: A Handbook for Practice on a Scientific Basis; Medpharm, Scientific Publisher Stuttgart: Stuttgart, Germany, 2004. [Google Scholar]
- European Pharmacopoeia Council of Europe. European Pharmacopoeia, 8th ed.; European Pharmacopoeia Council of Europe: Strasbourg, France, 2013. [Google Scholar]
- Agencia Nacional de Vigilancia Sanitaria y Fundación Oswaldo Cruz/Editora. Farmacopea Brasileña, 5th ed.; Agencia Nacional de Vigilancia Sanitaria y Fundación Oswaldo Cruz/Editora: Brasilia, Brazil, 2010. [Google Scholar]
- United States Pharmacopeia (USP 30), NF, 25th ed.; The United States Pharmacopeial Convention Inc.: Rockville, MD, USA, 2007.
- World Health Organization (WHO). Quality Control Methods for Medicinal Plant Materials; WHO: Geneva, Switzerland, 1998. [Google Scholar]
- World Health Organization (WHO). Quality Control Methods for Herbal Materials; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Araújo, E.R.D.; Félix-Silva, J.; Xavier-Santos, J.B.; Fernandes, J.M.; Guerra, G.C.B.; de Araújo, A.A.; Zucolotto, S.M. Local anti-inflammatory activity: Topical formulation containing Kalanchoe brasiliensis and Kalanchoe pinnata leaf aqueous extract. Biomed. Pharmacother. 2019, 113, 108721. [Google Scholar] [CrossRef] [PubMed]
- Palaniselvam, K.; Kyung, D.; Chae, E.S.; Soundharrajan, I.; Srisesharam, S.; Mariadhas, V.A.; Ki Choon, C. Quantification of major phenolic and flavonoid markers in forage crop Lolium multiflorum using HPLC-DAD. Rev. Bras. Farmacogn. 2018, 28, 282–288. [Google Scholar]
- Morais, J.; Ortiz, S.; Padilha, R.; Mandova, T.; Rodrigues, E.; Wilbur, A.; Michel, S.; Grougnet, R.; Zucolotto, S. Bryophyllum pinnatum markers: CPC isolation, simultaneous quantification by a validated UPLC-DAD method and biological evaluations. J. Pharm. Biomed. Anal. 2021, 193, 113682. [Google Scholar]
- Castro, E. Evaluación de Indicadores para la Diferenciación de Mieles Provenientes de la Zona Cafetera de la Sierra Nevada de Santa Marta. Master’s Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2015. [Google Scholar]
- Marino, O.; González, L.; Cortes, Y.; Camargo, J. Caracterización fitoquímica, determinación del contenido de lignina y la actividad antioxidante de los culmos de Guadua angustifolia Kunth. Rev. Fac. Cienc. Básicas 2015, 11, 124–135. [Google Scholar]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Validation of Analytical Procedures: Text and Methodology Q2(R1); International Conference on Harmonization: London, UK, 2005. [Google Scholar]
- Zeraik, M.L.; Yariwake, J.H. Quantification of isoorientin and total flavonoids in Passiflora edulis fruit pulpby HPLC-UV/DAD. Microchem. J. 2010, 96, 86–91. [Google Scholar] [CrossRef]
- Singh, S.; Bakshi, M. Guidance on Conduct of Stress Tests to Determine Inherent Stability of Drugs. Pharm. Technolology 2000, 4, 1–14. [Google Scholar]
- Food and Drug Administration (FDA). Guidance for Industry Drug Stability Guidelines; Center for Veterinary Medicine (CVM): Rockville, MD, USA, 2008. [Google Scholar]
- Comas, R. Contribución a la Estandarización del Proceso de Obtención a Escala de Laboratorio de un Extracto de las Hojas de Psidium guajava L. Master’s Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2014. [Google Scholar]
- Curtis, H.; Barnes, S.; Schnek, A.; Massarini, A. Biología, 7th ed.; Editorial Medica Panamericana: Madrid, España, 2008. [Google Scholar]
- Álvarez, N.; Bagué, A. Tecnología Farmacéutica; Editorial Club Universitario: Alicante, España, 2012. [Google Scholar]
- Bayona, A.; Peña, D. Evaluación Farmacognóstica y Antioxidante “In Vitro” del Extracto Etanólico de la Hoja del Aire (Kalanchoe pinnata). Master’s Thesis, Universidad de Guayaquil, Guayaquil, Ecuador, 2017. [Google Scholar]
- Camacho, O. Contribución a la Estandarización del Proceso de Obtención de un Extracto de Hojas de Petiveria alliacea l. (anamú) a Escala de Laboratorio. Master’s Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2014. [Google Scholar]
- Dewick, P.M. Medicinal Natural Products: A Biosynthetic Approach, 3rd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Li, Z.H.; Guo, H.; Xu, W.B.; Ge, J.; Li, X.; Alimu, M.; He, D.J. Rapid Identification of Flavonoid Constituents Directly from PTP1B Inhibitive Extract of Raspberry (Rubus idaeus L.) Leaves by HPLC-ESI QTOF-MS-MS. J. Chromatogr. Sci. 2016, 54, 805–810. [Google Scholar] [CrossRef]
- García, P.; Ayuso, M.; Lozano, E.; Pereira, C.; Dias, M.; Ivanov, M.; Calhelha, R.; Soković, M.; Ferreira, I.; Barros, L.; et al. Phenolic profiling and in vitro bioactivities of three medicinal Bryophyllum plants. Ind. Crop. Prod. 2021, 162, 113241. [Google Scholar] [CrossRef]
- Saldanha, L.; Vilegas, W.; Dokkedal, A. Characterization of flavonoids and phenolic acids in Myrcia bella Cambess. using FIA-ESI-IT-MS(n) and HPLC-PAD-ESI-IT-MS combined with NMR. Molecules 2013, 18, 8402–8416. [Google Scholar] [CrossRef] [PubMed]
- Roriz, C.L.; Barros, L.; Carvalho, A.M.; Santos-Buelga, C.; Ferreira, I.C.F.R. Pterospartum tridentatum, Gomphrena globosa and Cymbopogon citratus: A phytochemical study focused on antioxidant compounds. Food Res. Int. 2014, 62, 684–693. [Google Scholar] [CrossRef]
- Chen, G.; Li, X.; Saleri, F.; Guo, M. Analysis of Flavonoids in Rhamnus davurica and Its Antiproliferative Activities. Molecules 2016, 21, 1275. [Google Scholar] [CrossRef] [PubMed]
- Vijayalakshmi, A.; Madhira, G. Anti-psoriatic activity of flavonoids from Cassia tora leaves using the rat ultraviolet B ray photodermatitis model. Rev. Bras. Farmacogn. 2014, 24, 322–329. [Google Scholar] [CrossRef]
- Brito, A.; Ramirez, J.E.; Areche, C.; Sepúlveda, B.; Simirgiotis, M.J. HPLC-UV-MS profiles of phenolic compounds and antioxidant activity of fruits from three citrus species consumed in Northern Chile. Molecules 2014, 19, 17400–17421. [Google Scholar] [CrossRef]
- Ibrahim, L.; Elkhateeb, A.; Marzouk, M.; Hussein, S.; Abdel-Hameed, E.L.; Kassem, M. Flavonoid investigation, LC–ESI-MS profile and cytotoxic activity of Raphanus raphanistrum L. (Brassicaceae). J. Chem. Pharm. Res. 2016, 8, 786–793. [Google Scholar]
- El Sayed, A.M.; Ezzat, S.M.; El Naggar, M.M.; El Hawary, S.S. In vivo diabetic wound healing effect and HPLC–DAD–ESI–MS/MS profiling of the methanol extracts of eight Aloe species. Rev. Bras. Farmacogn. 2016, 26, 352–362. [Google Scholar] [CrossRef]
- Costa, S.; Muzitano, M.; Camargo, L.; Coutinho, M.A. Therapeutic Potential of Kalanchoe Species: Flavonoids and other Secondary Metabolites. Nat. Prod. Commun. 2008, 3, 2151–2164. [Google Scholar] [CrossRef]
- Horwitz, W.; Albert, R.J. The Horwitz ratio (HorRat): A Useful Index of Method Performance with Respect to Precision. J. Assoc. Off. Anal. Chem. 2006, 89, 1095–1109. [Google Scholar] [CrossRef]
- Rodríguez, J.; Amaya, C.; Caballero, P.; Alanís, M.; Aguilera, C.; Báez, J.; Moreno, S.; Núñez, A. Optimización de un método para determinación de fitoesteroles y escualeno en ácidos grasos destilados usando la metodología de superficie de respuesta y su validación. Rev. Mex. Cienc. Farm. 2014, 45, 1–10. [Google Scholar]
- Paredes, F.; Clemente, A. Polifenoles de aplicación en farmacia. Ámbito Farm. Fitoter. 2005, 24, 85–94. [Google Scholar]
- Justesen, U.; Arrigoni, E.; Larsen, B.; Amado, R. Degradation of Flavonoid Glycosides and Aglycones During in vitro Fermentation with Human Faecal Flora. LWT Food Sci. Technol. 2000, 33, 424–430. [Google Scholar] [CrossRef]
- Vetrova, E.; Maksimenko, E.; Khizrieva, S.; Bugaeva, A.; Borisenko, N.; Minkin, V. A simple way for the preparation of natural antioxidant quercetin from rutin by subcritical water. J. Nat. Sci. Biol. Med. 2017, 8, 213–215. [Google Scholar] [PubMed]
- Fransina, E.; Tanasale, M.; Latupeirissa, J.; Malle, D.; Tahapary, R. Phytochemical screening of water extract of gayam (Inocarpus edulis) Bark and its amylase inhibitor activity assay. Mater. Sci. Eng. 2019, 509, 012074. [Google Scholar] [CrossRef]
- Dou, M.; He, X.; Sun, Y.; Peng, F.; Liu, J.; Hao, L.; Yang, S. Controlled acid hydrolysis and kinetics of flavone C-glycosides from trollflowers. Chin. Chem. Lett. 2015, 26, 255–258. [Google Scholar] [CrossRef]
- Mandalari, G.; Bennett, R.N.; Kirby, A.R.; Lo Curto, R.B.; Bisignano, G.; Waldron, K.W.; Faulds, C.B. Enzymatic hydrolysis of flavonoids and pectic oligosaccharides from bergamot (Citrus bergamia Risso) peel. J. Agric. Food. Chem. 2006, 54, 8307–8313. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Title 21 Chapter I—FDA Department of health and Human Services Subchapter C—DRUGS: GENERAL PART 205. Guidelines for state licensing of wholesale prescription drug distributors. In CFR—Code of Federal Regulations; FDA: Washington, DC, USA, 2020. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Percentage (%) |
|---|---|
| Foreign matter | 0.3 |
| Humidity (loss on drying) | 10.0 ± 0.1 |
| Particle size distribution | 70.0 |
| Total ashes | 11.8 ± 0.6 |
| Peak | Retention Time (min) | Theoretical Mass [MߝH]− | Calculated Mass [MߝH]− | Error (Δ ppm) | Compound | Reference |
|---|---|---|---|---|---|---|
| 1 | 5.3 | 609.5085 | 609.5096 | −1.74 | Kaempferol-3-O-glucose-glucose | [48] |
| 3 | 5.9 | 479.0831 | 479.0724 | 22.33 | Myricetin-3-O-hexoside | [49] |
| 4 | 6.1 | 449.0720 | 449.2013 | −287.93 | Unknow flavonol | [50] |
| 5a | 6.5 | 463.0882 | 463.0870 | 2.59 | Quercetin 3-O-galactoside (Hyperoside) | [48] |
| 5b | 6.5 | 595.1299 | 595.1290 | 1.51 | Quercetin 3-O-(6-pentosyl)-hexoside | [51] |
| 6 | 6.7 | 463.0882 | 463.0871 | 2.38 | Quercetin 3-O-glucoside (Isoquercitrin) | [52] |
| 7 | 6.9 | 477.0674 | 477.0664 | 2.10 | Quercetin 3-O-glucuronide | [48,53] |
| 8 | 7.1 | 433.0776 | 433.0759 | 3.93 | Quercetin-O-arabinofuranoside | [50] |
| 9 | 7.3 | 477.0942 | 477.0672 | 56.59 | Isoramnetin-3-O-hexoside | [54] |
| 10 | 7.7 | 579.1355 | 579.1347 | 1.38 | Quercetin 3-O-α-L-arabinopyranosyl (1→2) α-L-rhamnopyranoside | [7,18,55] |
| 11 | 7.9 | 447.0932 | 447.0921 | 2.46 | Quercetin 3-O-rhamnoside | [7,18] |
| 12 | 8.1 | 579.1355 | 579.1343 | 2.07 | Kaempferol-3-O-pentosyl-hexoside | [51,56] |
| 13 | 9.1 | 563.1406 | 563.1402 | 0.76 | Kaempferol 3-O-α-L-arabinopyranosyl-(1→2) α-L-rhamnopyranoside | [18] |
| 14 | 9.3 | 593.1511 | 593.1500 | 1.85 | Isoscoparin-7-O-arabinoside | [18] |
| Calibration Curve | Calibration Equation a | Correlation Factor (r2) | Linear Range (µg mL−1) |
|---|---|---|---|
| 1 | y = 12243x − 17431 | 0.9997 | 0.1953–400.0 |
| 2 | y = 12232x − 15714 | 0.9997 | 0.1953–400.0 |
| 3 | y = 12225x − 16640 | 0.9997 | 0.1953–400.0 |
| Repeatability b | Intermediate precision b | ||
| Rutin concentration (μg mL−1) | RSD (%) | RSD (%) | |
| 250.0 | 0.115 | 0.016 | |
| 50.0 | 0.181 | 0.025 | |
| 1.562 | 1.198 | 0.062 | |
| LOQ c (μg mL−1) | LOD c (μg mL−1) | ||
| Experimental value | 0.0976 | 0.0488 | |
| Theoric value | 0.0026 | 0.0009 |
| RSD (%) | Retention Time (min) | |
|---|---|---|
| Temperature (25 °C) | ||
| 0.801 | 6.6 | |
| Flow rate (0.3 mL min−1) | ||
| 0.467 | 6.8 | |
| Formic acid 0.5% | ||
| 0.869 | 6.3 |
| Collection | Content % a | mg EQ-Rutin/mg Extract |
|---|---|---|
| November 2019 | 12.48 ± 0.50 | 0.124 ± 0.005 |
| December 2020 | 9.97 ± 0.17 | 0.099 ± 0.001 |
| January 2021 | 8.51 ± 0.35 | 0.085 ± 0.008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villarreal Romero, W.L.; Robles Camargo, J.E.; Costa, G.M. Phytochemical Standardization of an Extract Rich in Flavonoids from Flowers of Kalanchoe pinnata (Lam) Pers. Sci. Pharm. 2023, 91, 50. https://doi.org/10.3390/scipharm91040050
Villarreal Romero WL, Robles Camargo JE, Costa GM. Phytochemical Standardization of an Extract Rich in Flavonoids from Flowers of Kalanchoe pinnata (Lam) Pers. Scientia Pharmaceutica. 2023; 91(4):50. https://doi.org/10.3390/scipharm91040050
Chicago/Turabian StyleVillarreal Romero, Wilson Leonardo, Jorge Eliecer Robles Camargo, and Geison Modesti Costa. 2023. "Phytochemical Standardization of an Extract Rich in Flavonoids from Flowers of Kalanchoe pinnata (Lam) Pers" Scientia Pharmaceutica 91, no. 4: 50. https://doi.org/10.3390/scipharm91040050
APA StyleVillarreal Romero, W. L., Robles Camargo, J. E., & Costa, G. M. (2023). Phytochemical Standardization of an Extract Rich in Flavonoids from Flowers of Kalanchoe pinnata (Lam) Pers. Scientia Pharmaceutica, 91(4), 50. https://doi.org/10.3390/scipharm91040050