Integrated Metabolomics and Transcriptomics Suggest the Global Metabolic Response to 2-Aminoacrylate Stress in Salmonella enterica


 and
and
Abstract
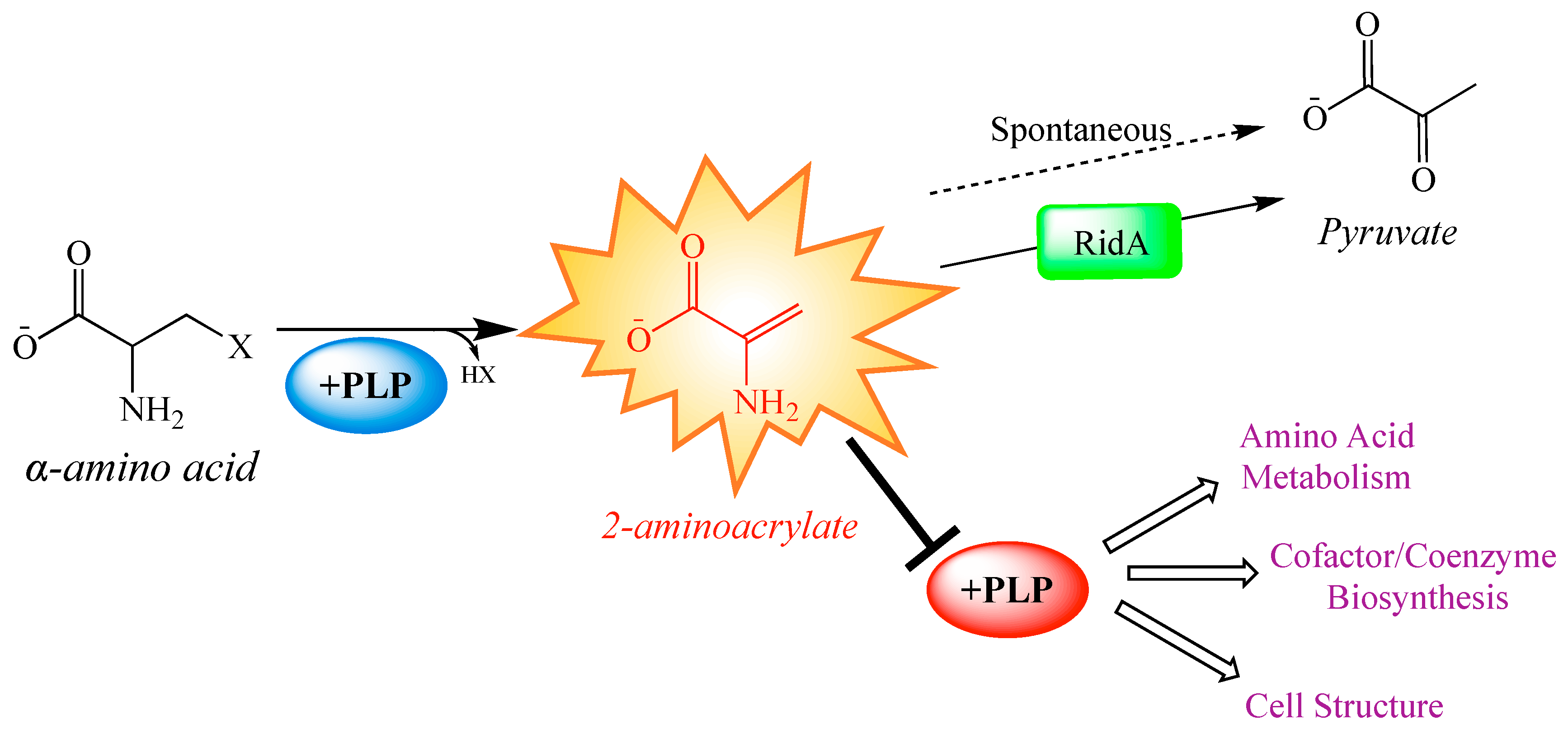
1. Introduction
2. Results and Discussion
2.1. Metabolite Levels Are Altered in an S. enterica RidA Mutant
2.2. Transcriptome Analyses of RidA Mutant Complements Metabolomics Data
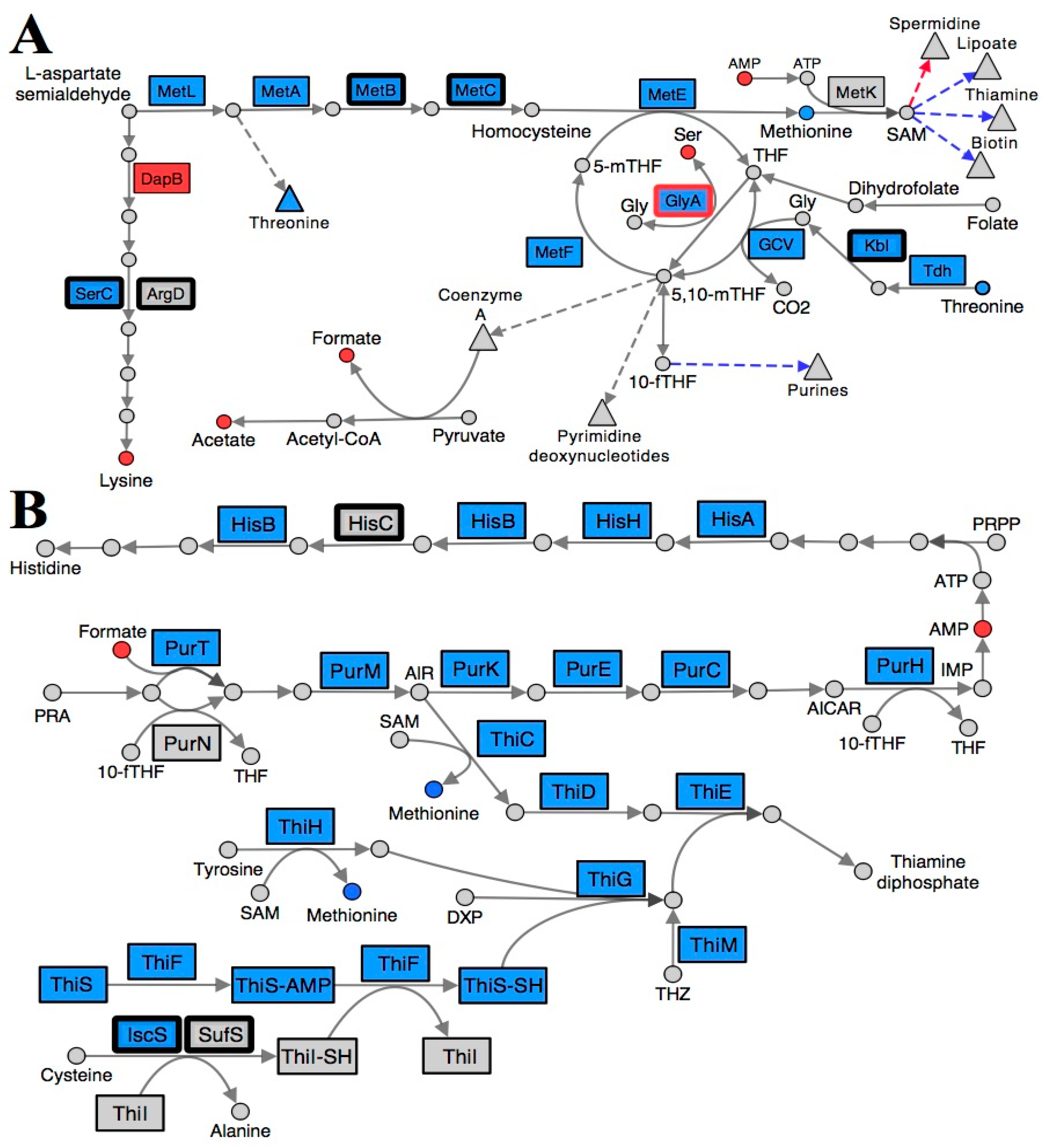
2.3. Folate Metabolism Is Perturbed in a RidA Mutant
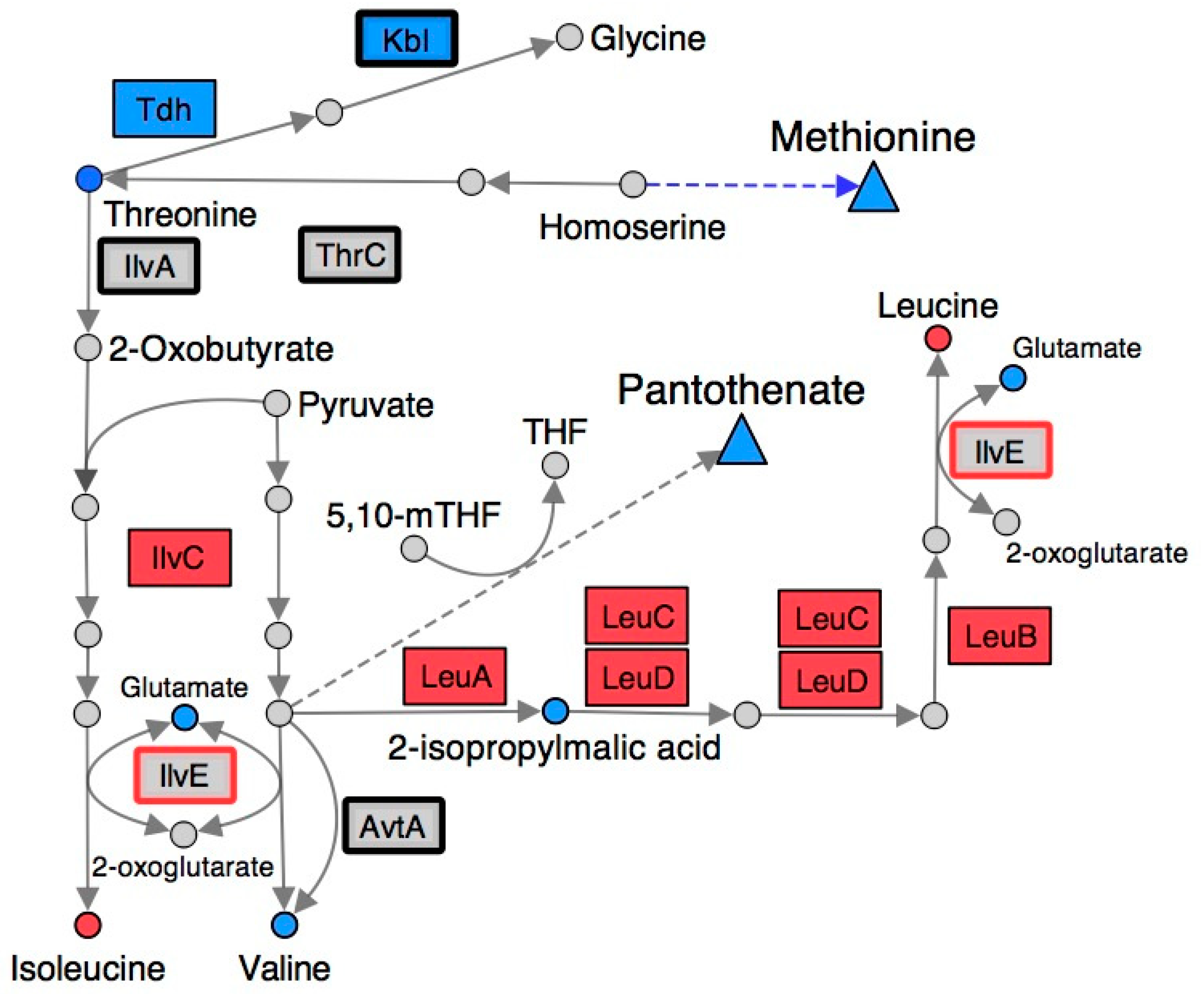
2.4. Branched-Chain Amino Acid (BCAA) Metabolism Is Altered in a RidA Mutant
2.5. Many Metabolic Perturbations Consistent with Transaminase Damage by 2AA
3. Materials and Methods
3.1. Bacterial Strains, Chemicals and Media
3.2. Metabolomics Cell Preparation
3.3. Metabolite Extraction
3.4. Acquisition and Processing of NMR Spectral Data
3.4.1. 1D 1H
3.4.2. J-RES
3.5. Compound Identification/Database Matching
3.6. Quantification of Metabolomics Data
3.7. Pathway Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Keller, M.A.; Piedrafita, G.; Ralser, M. The widespread role of non-enzymatic reactions in cellular metabolism. Curr. Opin. Biotechnol. 2015, 34, 153–161. [Google Scholar] [CrossRef]
- De Lorenzo, V.; Sekowska, A.; Danchin, A. Chemical reactivity drives spatiotemporal organisation of bacterial metabolism. FEMS Microbiol. Rev. 2015, 39, 96–119. [Google Scholar] [CrossRef]
- Albert, R.; Jeong, H.; Barabasi, A.-L. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Furlan, S.N.; Watkins, B.; Tkachev, V.; Flynn, R.; Cooley, S.; Ramakrishnan, S.; Singh, K.; Giver, C.; Hamby, K.; Stempora, L.; et al. Transcriptome analysis of GVHD reveals aurora kinase A as a targetable pathway for disease prevention. Sci. Transl. Med. 2015, 7, 315ra191. [Google Scholar] [CrossRef] [PubMed]
- Koenig, D.; Jimenez-Gomez, J.M.; Kimura, S.; Fulop, D.; Chitwood, D.H.; Headland, L.R.; Kumar, R.; Covington, M.F.; Devisetty, U.K.; Tat, A.V.; et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. USA 2013, 110, E2655–E2662. [Google Scholar] [CrossRef]
- Lee-McMullen, B.; Chrzanowski, S.M.; Vohra, R.; Forbes, S.C.; Vandenborne, K.; Edison, A.S.; Walter, G.A. Age-dependent changes in metabolite profile and lipid saturation in dystrophic mice. NMR Biomed. 2019, 32, e4075. [Google Scholar] [CrossRef]
- Clendinen, C.S.; Gaul, D.A.; Monge, M.E.; Arnold, R.S.; Edison, A.S.; Petros, J.A.; Fernández, F.M. Preoperative metabolic signatures of prostate cancer recurrence following radical prostatectomy. J. Proteome Res. 2019, 18, 1316–1327. [Google Scholar] [CrossRef]
- Vincent, I.M.; Ehmann, D.E.; Mills, S.D.; Perros, M.; Barrett, M.P. Untargeted metabolomics to ascertain antibiotic modes of action. Antimicrob. Agents Chemother. 2016, 60, 2281–2291. [Google Scholar] [CrossRef]
- Mareya, C.; Tugizimana, F.; Piater, L.; Madala, N.; Steenkamp, P.; Dubery, I. Untargeted metabolomics reveal defensome-related metabolic reprogramming in Sorghum bicolor against infection by Burkholderia andropogonis. Metabolites 2019, 9, 8. [Google Scholar] [CrossRef]
- Hattori, A.; Tsunoda, M.; Konuma, T.; Kobayashi, M.; Nagy, T.; Glushka, J.; Tayyari, F.; McSkimming, D.; Kannan, N.; Tojo, A.; et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017, 545, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Baptista, R.; Fazakerley, D.M.; Beckmann, M.; Baillie, L.; Mur, L.A.J. Untargeted metabolomics reveals a new mode of action of pretomanid (PA-824). Sci. Rep. 2018, 8, 5084. [Google Scholar] [CrossRef] [PubMed]
- Downs, D.M.; Bazurto, J.V.; Gupta, A.; Fonseca, L.L.; Voit, E.O. The three-legged stool of understanding metabolism: Integrating metabolomics with biochemical genetics and computational modeling. AIMS Microbiol. 2018, 4, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Bazurto, J.V.; Dearth, S.P.; Tague, E.D.; Campagna, S.R.; Downs, D.M. Untargeted metabolomics confirms and extends the understanding of the impact of aminoimidazole carboxamide ribotide (AICAR) in the metabolic network of Salmonella enterica. Microb. Cell 2018, 5, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, J.A.; Flynn, J.M.; Downs, D.M. Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5’-phosphate (PLP)-dependent enzyme reactions. J. Biol. Chem. 2012, 287, 3454–3461. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, T.D.; Nguyen, T.N.D.; Gidda, S.K.; ElBadawi-Sidhu, M.; Lambrecht, J.A.; McCarty, D.R.; Downs, D.M.; Cooper, A.J.L.; Fiehn, O.; Mullen, R.T.; et al. Arabidopsis and maize RidA proteins preempt reactive enamine/imine damage to branched-chain amino acid biosynthesis in plastids. Plant Cell 2014, 26, 3010–3022. [Google Scholar] [CrossRef]
- Ernst, D.C.; Lambrecht, J.A.; Schomer, R.A.; Downs, D.M. Endogenous synthesis of 2-aminoacrylate contributes to cysteine sensitivity in Salmonella enterica. J. Bacteriol. 2014, 196, 3335–3342. [Google Scholar] [CrossRef]
- Hodge-Hanson, K.M.; Downs, D.M. Members of the Rid protein family have broad imine deaminase activity and can accelerate the Pseudomonas aeruginosa D-arginine dehydrogenase (DauA) reaction in vitro. PLoS ONE 2017, 12, e0185544. [Google Scholar] [CrossRef]
- Ernst, D.C.; Downs, D.M. Mmf1p couples amino acid metabolism to mitochondrial DNA maintenance in Saccharomyces cerevisiae. MBio 2018, 9. [Google Scholar] [CrossRef]
- Degani, G.; Barbiroli, A.; Regazzoni, L.; Popolo, L.; Vanoni, M.A. Imine deaminase activity and conformational stability of UK114, the mammalian member of the Rid protein family active in amino acid metabolism. Int. J. Mol. Sci. 2018, 19, 945. [Google Scholar] [CrossRef]
- Lambrecht, J.A.; Schmitz, G.E.; Downs, D.M. RidA proteins prevent metabolic damage inflicted by PLP-dependent dehydratases in all domains of life. MBio 2013, 4, e00033-13. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Christopherson, M.R.; Downs, D.M. Decreased coenzyme A levels in ridA mutant strains of Salmonella enterica result from inactivated serine hydroxymethyltransferase: ridA mutants are deficient in one carbon metabolism. Mol. Microbiol. 2013, 89, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Downs, D.M. In the absence of RidA, endogenous 2-aminoacrylate inactivates alanine racemases by modifying the pyridoxal 5’-phosphate cofactor. J. Bacteriol. 2013, 195, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Borchert, A.J.; Downs, D.M. The response to 2-aminoacrylate differs in Escherichia coli and Salmonella enterica, despite shared metabolic components. J. Bacteriol. 2017, 199, e00140-17. [Google Scholar] [CrossRef]
- Ernst, D.C.; Downs, D.M. 2-aminoacrylate stress induces a context-dependent glycine requirement in ridA strains of Salmonella enterica. J. Bacteriol. 2016, 198, 536–543. [Google Scholar] [CrossRef]
- Percudani, R.; Peracchi, A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003, 4, 850–854. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Borchert, A.J.; Downs, D.M. Endogenously generated 2-aminoacrylate inhibits motility in Salmonella enterica. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Downs, D.M.; Ernst, D.C. From microbiology to cancer biology: The Rid protein family prevents cellular damage caused by endogenously generated reactive nitrogen species: RidA stress response. Mol. Microbiol. 2015, 96, 211–219. [Google Scholar] [CrossRef]
- Borchert, A.J.; Ernst, D.C.; Downs, D.M. Reactive enamines and imines in vivo: Lessons from the RidA paradigm. Trends Biochem. Sci. 2019, 44, 849–860. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Schmitz, G.E.; Downs, D.M. YjgF is required for isoleucine biosynthesis when Salmonella enterica is grown on pyruvate medium. J. Bacteriol. 2008, 190, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Krueger, J.H.; Itoh, S.; Deboer, H.A.; Nomura, M. Growth-rate-dependent regulation of ribosome synthesis in Escherichia coli—Expression of the Iacz and galK genes fused to ribosomal promoters. Cell 1981, 25, 773–782. [Google Scholar] [CrossRef]
- Aseev, L.V.; Koledinskaya, L.S.; Boni, I.V. Regulation of ribosomal protein operons rplM-rpsI, rpmB-rpmG, and rplU-rpmA at the transcriptional and translational levels. J. Bacteriol. 2016, 198, 2494–2502. [Google Scholar] [CrossRef]
- Lemke, J.J.; Sanchez-Vazquez, P.; Burgos, H.L.; Hedberg, G.; Ross, W.; Gourse, R.L. Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc. Natl. Acad. Sci. USA 2011, 108, 5712–5717. [Google Scholar] [CrossRef]
- Lucock, M. Folic acid: Nutritional biochemistry, molecular biology, and role in disease processes. Mol. Genet. Metab. 2000, 71, 121–138. [Google Scholar] [CrossRef]
- White, R.J.; Phillips, D.R. Bidirectional transcription footprinting of DNA-binding ligands. Biochemistry 1989, 28, 6259–6269. [Google Scholar] [CrossRef]
- Webb, M. Pyruvate accumulation in growth-inhibited cultures of Aerobacter aerogenes. Biochem. J. 1968, 106, 375–380. [Google Scholar] [CrossRef]
- Webb, M. Aminopterin inhibition in Aerobacter aerogenes: Alanine and valine accumulation during the inhibition and their utilization on recovery. Biochem. J. 1958, 70, 472–489. [Google Scholar] [CrossRef]
- Lal, P.B.; Schneider, B.L.; Vu, K.; Reitzer, L. The redundant aminotransferases in lysine and arginine synthesis and the extent of aminotransferase redundancy in Escherichia coli: Aminotransferase redundancy. Mol. Microbiol. 2014, 94, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pittard, J. Biosynthesis of the aromatic amino acids. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Ravnikar, P.D.; Somerville, R.L. Genetic characterization of a highly efficient alternate pathway of serine biosynthesis in Escherichia coli. J. Bacteriol. 1987, 169, 7. [Google Scholar] [CrossRef] [PubMed]
- Castilho, B.A.; Olfson, P.; Casadaban, M.J. Plasmid insertion mutagenesis and lac gene fusion with mini-mu bacteriophage transposons. J. Bacteriol. 1984, 158, 488–495. [Google Scholar]
- Schmitz, G.; Downs, D.M. Reduced transaminase B (IlvE) activity caused by the lack of yjgF is dependent on the status of threonine deaminase (IlvA) in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2004, 186, 803–810. [Google Scholar] [CrossRef]
- Vogel, H.J.; Bonner, D.M. Acetylornithinase of Escherichia coli: Partial purification and some properties. J. Biol. Chem. 1956, 218, 97–106. [Google Scholar]
- Balch, W.E.; Wolfe, R.S. New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressureized atmosphere. Appl. Environ. Microbiol. 1976, 32, 781–791. [Google Scholar]
- Sakhaii, P.; Bermel, W. Improving the sensitivity of conventional spin echo spectra by preservation of initial signal-to-noise ratio. J. Magn. Reson. 2014, 242, 220–223. [Google Scholar] [CrossRef]
- Bingol, K.; Li, D.W.; Zhang, B.; Bruschweiler, R. Comprehensive metabolite identification strategy using multiple two-dimensional NMR spectra of a complex mixture implemented in the COLMARm web server. Anal. Chem. 2016, 88, 12411–12418. [Google Scholar] [CrossRef]
- Walejko, J.M.; Chelliah, A.; Keller-Wood, M.; Gregg, A.; Edison, A.S. Global metabolomics of the placenta reveals distinct metabolic profiles between maternal and fetal placental tissues following delivery in non-labored women. Metabolites 2018, 8, 10. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Enos-Berlage, J.L.; Langendorf, M.J.; Downs, D.M. Complex metabolic phenotypes caused by a mutation in yjgF, encoding a member of the highly conserved YER057c/YjgF family of proteins. J. Bacteriol. 1998, 180, 6519–6528. [Google Scholar] [PubMed]
- Lambrecht, J.A.; Browne, B.A.; Downs, D.M. Members of the YjgF/YER057c/UK114 family of proteins inhibit phosphoribosylamine synthesis in vitro. J. Biol. Chem. 2010, 285, 34401–34407. [Google Scholar] [CrossRef]
- Ernst, D.C.; Anderson, M.E.; Downs, D.M. L-2,3-diaminopropionate generates diverse metabolic stresses in Salmonella enterica: Targets of diaminopropionate in Salmonella. Mol. Microbiol. 2016, 101, 210–223. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| 1 Compound | 2 Mean (SD) | 3 Fold Change | p Value | FDR Value | |
|---|---|---|---|---|---|
| Wild-Type | ridA | ||||
| Valine | 1.23 (0.13) | 2.71 (0.13) | 2.20 | 8.47 × 10−14 | 3.56× 10−12 |
| Methionine | 0.08 (0.01) | 0.17 (0.01) | 2.20 | 4.43 × 10−12 | 9.31× 10−11 |
| GSSG | 0.54 (0.20) | 2.02 (0.18) | 3.71 | 1.99× 10−11 | 2.78× 10−10 |
| CMP | 0.05 (0.02) | 0.13 (0.02) | 2.94 | 1.38 × 10−8 | 8.29 × 10−8 |
| 2-isopropylmalic acid | 0.32 (0.04) | 1.30 (0.45) | 4.03 | 7.54× 10−6 | 3.52 × 10−5 |
| Glucose | 0.04 (0.01) | 0.06 (0.01) | 1.40 | 1.41 × 10−5 | 5.37 × 10−5 |
| Maltose | 1.41 (0.11) | 1.60 (0.10) | 1.13 | 2.13 × 10−3 | 5.17 × 10−3 |
| UTP | 0.04 (0.01) | 0.06 (0.01) | 1.43 | 8.79 × 10−3 | 0.02 |
| Glutamate | 0.18 (0.04) | 0.25 (0.06) | 1.41 | 9.82 × 10−3 | 0.02 |
| Threonine | 0.05 (0.01) | 0.06 (0.01) | 1.22 | 0.02 | 0.03 |
| Pantothenic acid | 0.13 (0.02) | 0.16 (0.02) | 1.17 | 0.03 | 0.04 |
| Leucine | 1.90 (0.25) | 0.80 (0.06) | 0.42 | 6.61 × 10−10 | 6.94 × 10−9 |
| Lysine | 0.59 (0.13) | 0.07 (0.02) | 0.12 | 1.53 × 10−9 | 1.29 × 10−8 |
| Phenylalanine | 0.13 (0.03) | 0.02 (0.01) | 0.12 | 1.38 × 10−8 | 8.29 × 10−8 |
| dCMP | 0.68 (0.08) | 0.44 (0.03) | 0.65 | 4.88 × 10−7 | 2.56 × 10−6 |
| Succinic acid | 2.49 (0.35) | 1.67 (0.20) | 0.67 | 1.39 × 10−5 | 5.37 × 10−5 |
| AMP | 0.70 (0.14) | 0.42 (0.05) | 0.60 | 2.92 × 10−5 | 1.02 × 10−4 |
| NADP | 0.05 (0.01) | 0.04 (0.01) | 0.74 | 9.71 × 10−5 | 3.14 × 10−4 |
| Formate | 0.16 (0.02) | 0.12 (0.02) | 0.76 | 2.79 × 10−4 | 8.38 × 10−4 |
| N-acetylputrescine | 0.16 (0.02) | 0.12 (0.02) | 0.77 | 1.11 × 10−3 | 3.10 × 10−3 |
| Isoleucine | 0.37 (0.12) | 0.22 (0.02) | 0.61 | 1.89 × 10−3 | 4.95 × 10−3 |
| Acetate | 2.50 (0.26) | 2.12 (0.18) | 0.85 | 2.22 × 10−3 | 5.17 × 10−3 |
| Serine | 0.06 (0.01) | 0.05 (0.01) | 0.87 | 0.01 | 0.03 |
| 1 Compound | 2 Mean (SD) | 3 Fold Change | p Value | FDR Value | |
|---|---|---|---|---|---|
| Wild-Type | ridA | ||||
| N-acetyl alanine | 0.02 (0.01) | 0.02 (0.01) | 1.24 | 0.07 | 0.10 |
| Alanine | 0.58 (0.12) | 0.68 (0.11) | 1.17 | 0.09 | 0.12 |
| CDP | 0.01 (0.01) | 0.02 (0.01) | 1.36 | 0.12 | 0.15 |
| Uracil | 0.06 (0.02) | 0.07 (0.03) | 1.25 | 0.22 | 0.27 |
| Ethanolamine | 0.04 (0.01) | 0.04 (0.02) | 1.15 | 0.45 | 0.53 |
| NMN | 0.34 (0.08) | 0.36 (0.07) | 1.05 | 0.61 | 0.68 |
| Tyrosine | 0.04 (0.02) | 0.04 (0.01) | 1.06 | 0.66 | 0.69 |
| 2-oxoglutaric acid | 0.08 (0.12) | 0.09 (0.16) | 1.05 | 0.95 | 0.95 |
| Malic acid | 0.04 (0.01) | 0.03 (0.01) | 0.88 | 0.05 | 0.07 |
| Putrescine | 0.47 (0.21) | 0.34 (0.15) | 0.72 | 0.15 | 0.19 |
| UMP | 0.19 (0.04) | 0.17 (0.02) | 0.93 | 0.36 | 0.43 |
| CTP | 0.21 (0.04) | 0.20 (0.03) | 0.95 | 0.57 | 0.65 |
| NAD | 0.09 (0.04) | 0.08 (0.03) | 0.92 | 0.64 | 0.69 |
| 2-aminobutyric acid | 1.17 (0.45) | 1.14 (0.09) | 0.97 | 0.84 | 0.86 |
| Pathway Description | 1 Relative Abundance | Differential Genes/Total Genes | p Value | FDR Value |
|---|---|---|---|---|
| Thiamine metabolism (stm00730) | ridA | 8/12 | 8.60 × 10−7 | 4.80 × 10−5 |
| Biotin metabolism (stm00780) | ridA | 6/14 | 6.60 × 10−4 | 1.20 × 10−2 |
| Biosynthesis of amino acids (stm01230) | ridA | 18/130 | 5.50 × 10−4 | 1.50 × 10−3 |
| One-carbon pool by folate (stm00670) | ridA | 5/13 | 4.40 × 10−3 | 4.90 × 10−2 |
| ABC transporters (stm02010) | ridA | 18/173 | 1.30 × 10−2 | 8.40 × 10−2 |
| Glycine, serine, and threonine metabolism (stm00260) | ridA | 7/35 | 1.10 × 10−2 | 8.70 × 10−2 |
| Flagellar assembly (stm02040) | WT | 31/37 | 3.00 × 10−28 | 1.40 × 10−26 |
| Ribosome (stm03010) | WT | 28/78 | 4.20 × 10−12 | 1.00 × 10−10 |
| Bacterial chemotaxis (stm02030) | WT | 11/23 | 3.60 × 10−6 | 5.70 × 10−5 |
| Bacterial invasion of epithelial cells (stm05100) | WT | 5/9 | 3.60 × 10−3 | 4.20 × 10−2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borchert, A.J.; Walejko, J.M.; Guennec, A.L.; Ernst, D.C.; Edison, A.S.; Downs, D.M. Integrated Metabolomics and Transcriptomics Suggest the Global Metabolic Response to 2-Aminoacrylate Stress in Salmonella enterica. Metabolites 2020, 10, 12. https://doi.org/10.3390/metabo10010012
Borchert AJ, Walejko JM, Guennec AL, Ernst DC, Edison AS, Downs DM. Integrated Metabolomics and Transcriptomics Suggest the Global Metabolic Response to 2-Aminoacrylate Stress in Salmonella enterica. Metabolites. 2020; 10(1):12. https://doi.org/10.3390/metabo10010012
Chicago/Turabian StyleBorchert, Andrew J., Jacquelyn M. Walejko, Adrien Le Guennec, Dustin C. Ernst, Arthur S. Edison, and Diana M. Downs. 2020. "Integrated Metabolomics and Transcriptomics Suggest the Global Metabolic Response to 2-Aminoacrylate Stress in Salmonella enterica" Metabolites 10, no. 1: 12. https://doi.org/10.3390/metabo10010012
APA StyleBorchert, A. J., Walejko, J. M., Guennec, A. L., Ernst, D. C., Edison, A. S., & Downs, D. M. (2020). Integrated Metabolomics and Transcriptomics Suggest the Global Metabolic Response to 2-Aminoacrylate Stress in Salmonella enterica. Metabolites, 10(1), 12. https://doi.org/10.3390/metabo10010012