A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics

 ,
, 

 , ,
, , 
Abstract
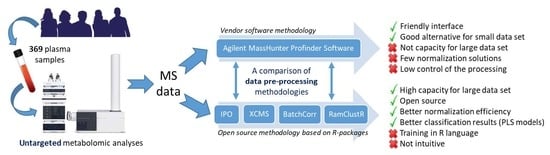
:
1. Introduction
2. Results and Discussion
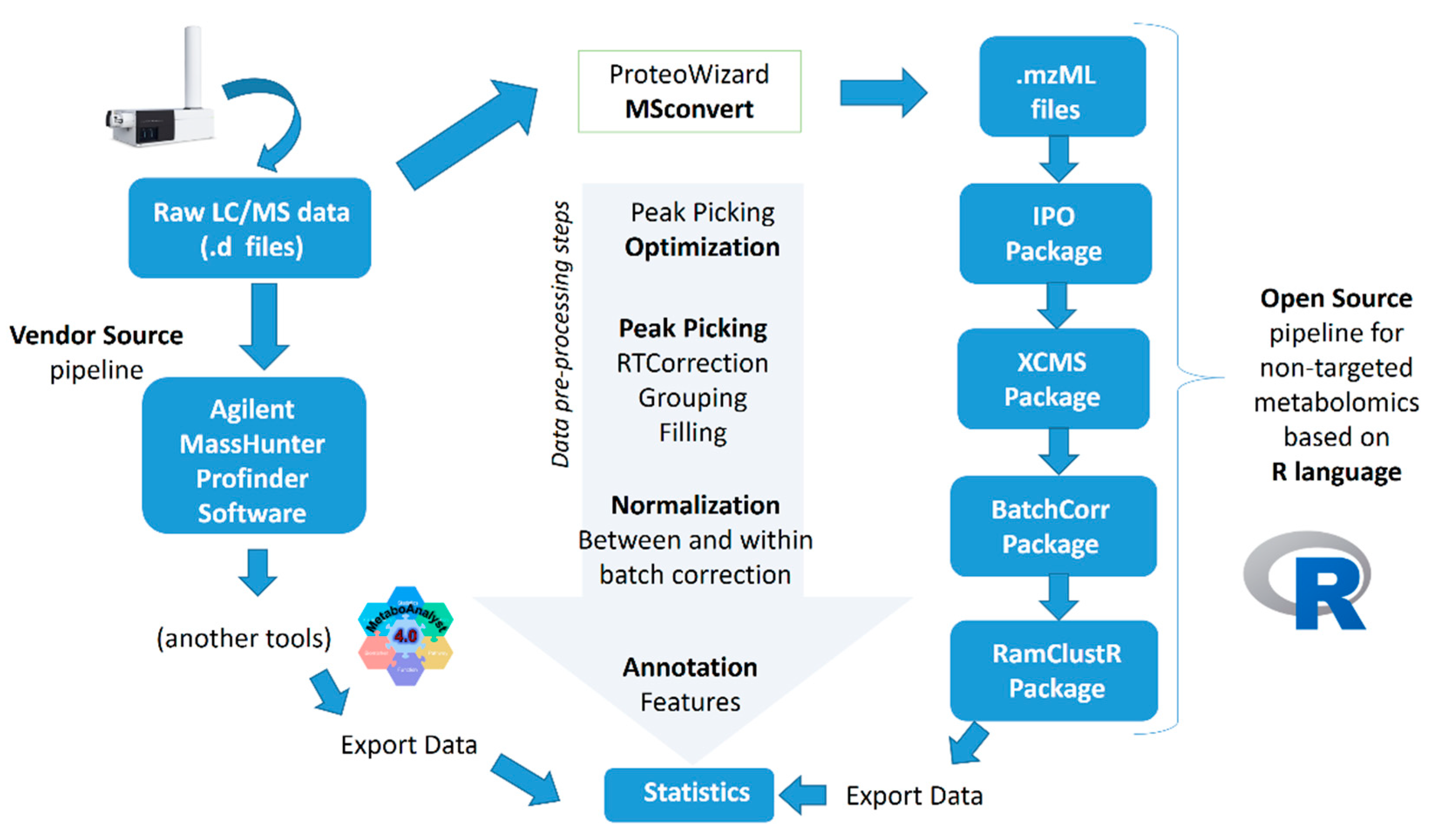
2.1. Peak Picking
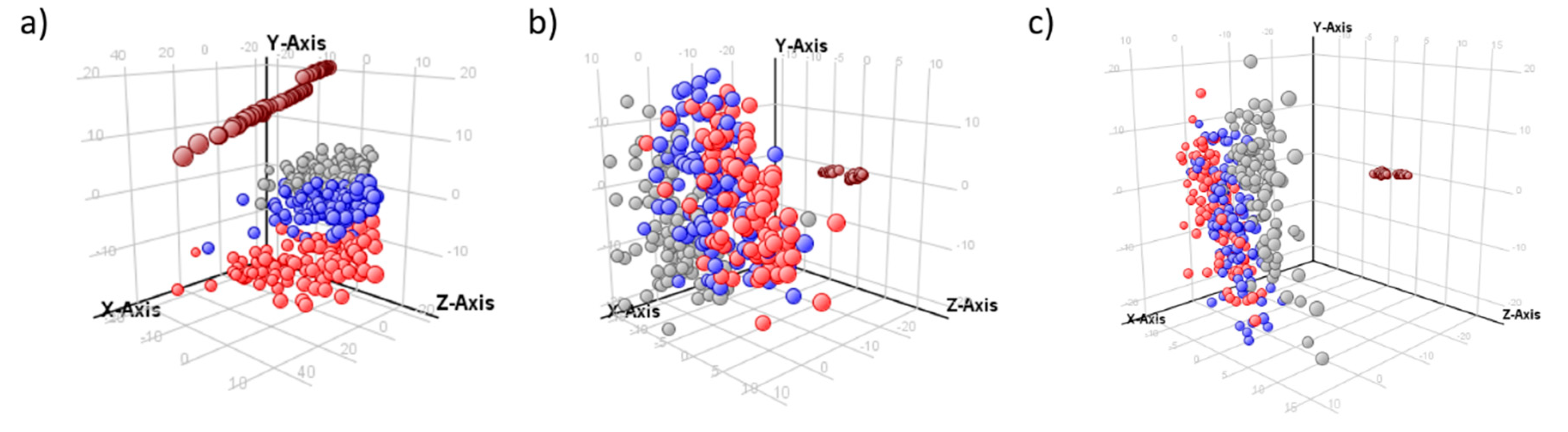
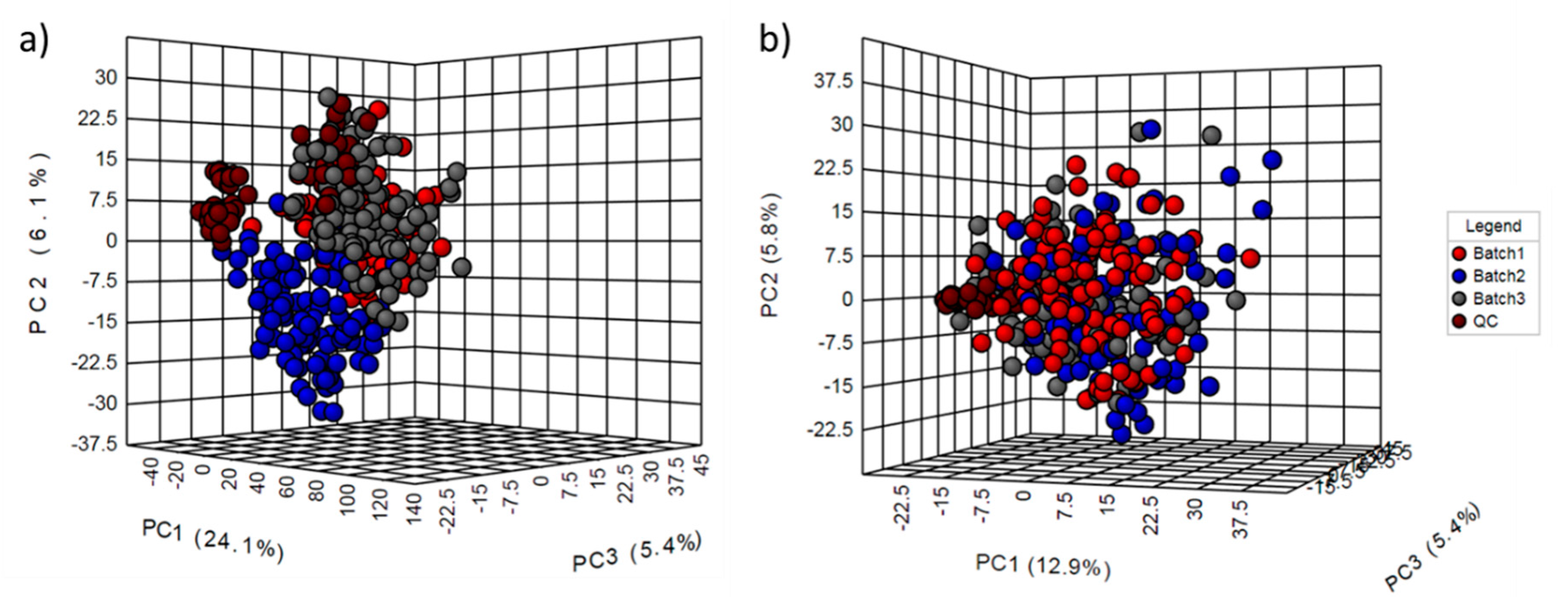
2.2. Normalization Results
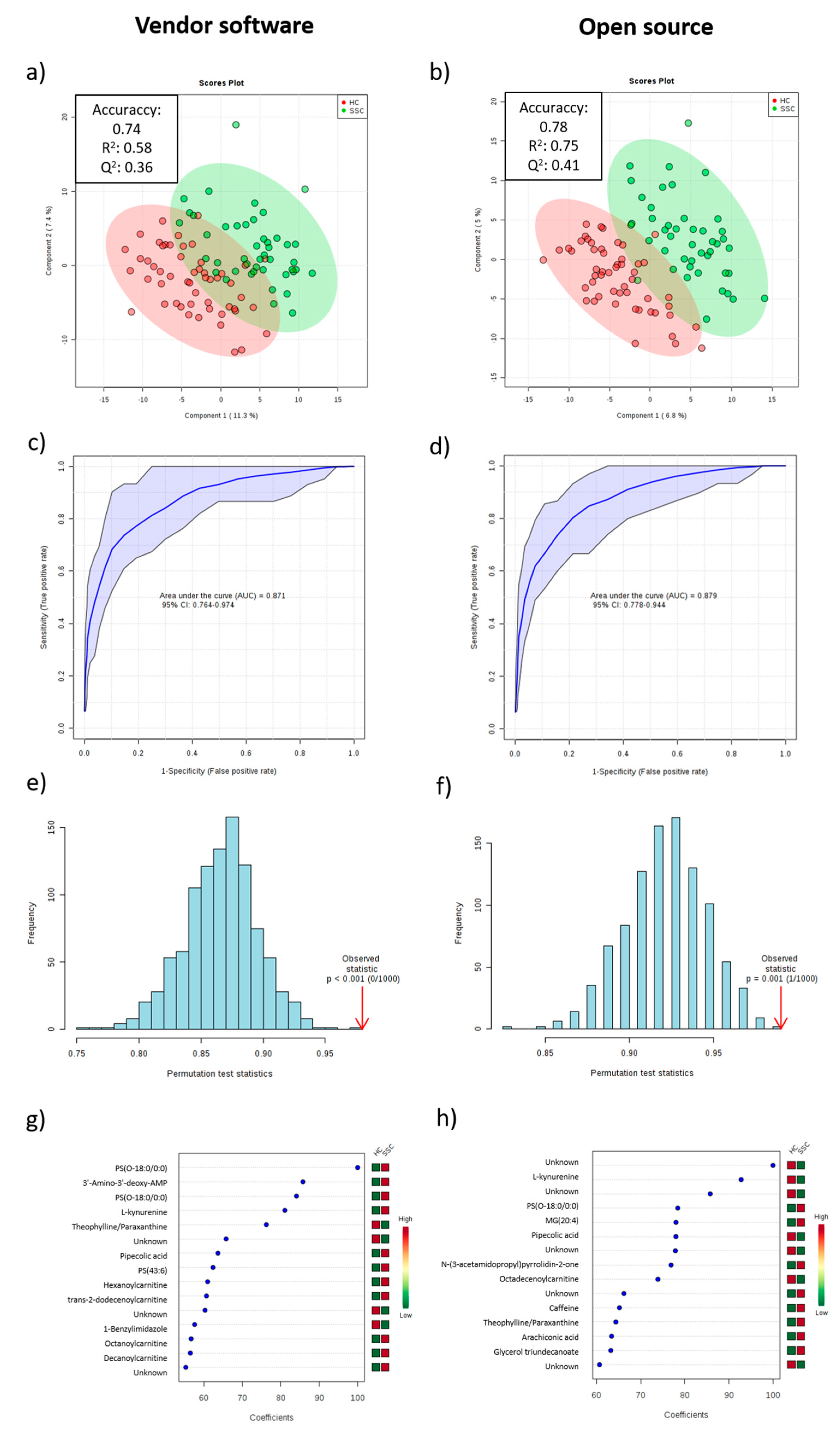
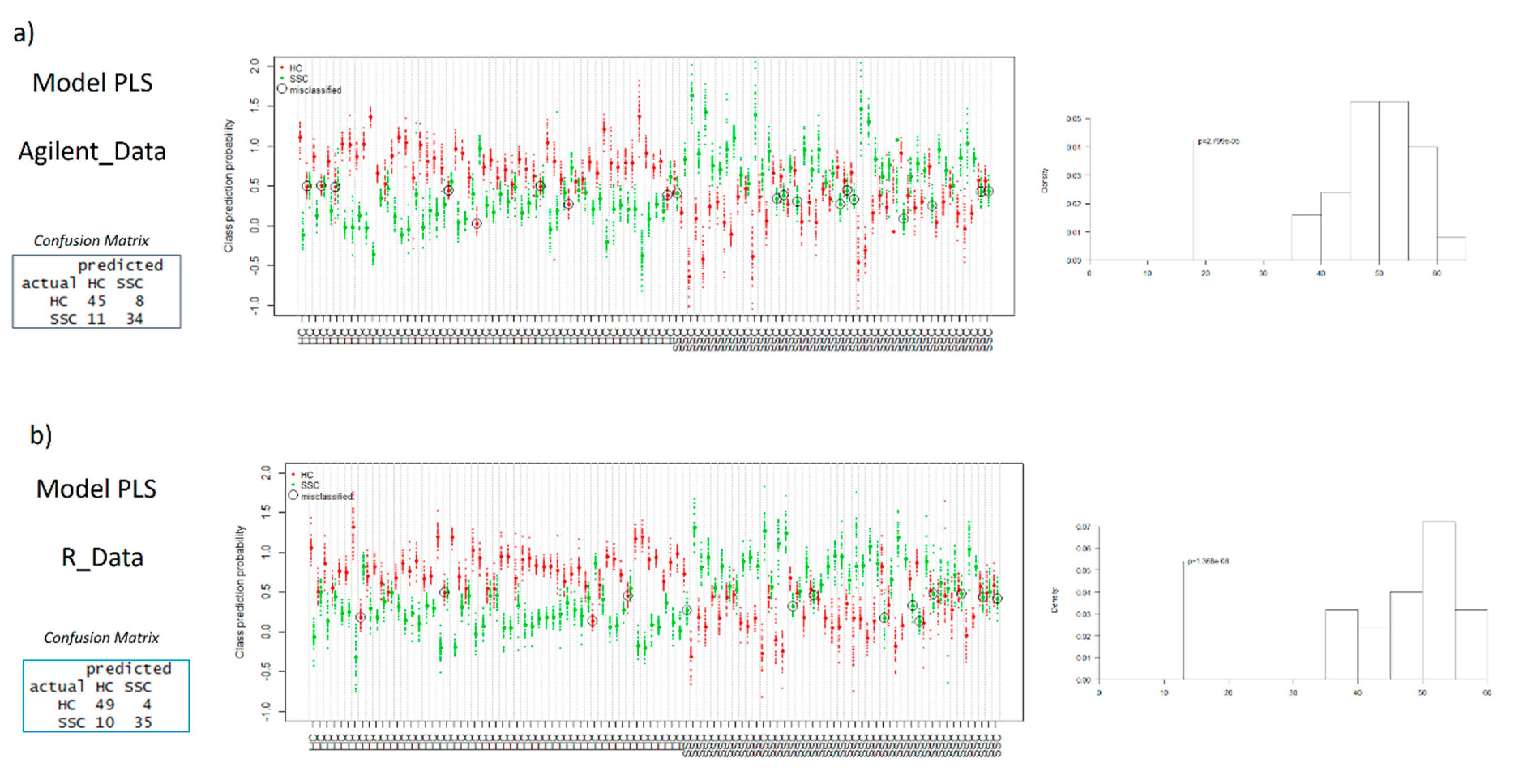
2.3. Multivariate Models
2.4. Global Comparison of Both Methodologies
3. Materials and Methods
3.1. Dataset
3.2. Data Pre-Processing
3.2.1. Agilent MassHunter Profinder Software Approach
3.2.2. R-Based Approach
3.3. Statistics and Metabolite Annotation
4. Conclusions and Future Research
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Agin, A.; Heintz, D.; Ruhland, E.; Chao de la Barca, J.M.; Zumsteg, J.; Moal, V.; Gauchez, A.S.; Namer, I.J. Metabolomics—An overview. From basic principles to potential biomarkers (part 1). Med. Nucl. 2016, 40, 4–10. [Google Scholar] [CrossRef]
- Parfieniuk, E.; Zbucka-Kretowska, M.; Ciborowski, M.; Kretowski, A.; Barbas, C. Untargeted metabolomics: An overview of its usefulness and future potential in prenatal diagnosis. Expert Rev. Proteom. 2018, 15, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulaszewska, M.M.; Weinert, C.H.; Trimigno, A.; Portmann, R.; Andres Lacueva, C.; Badertscher, R.; Brennan, L.; Brunius, C.; Bub, A.; Capozzi, F.; et al. Nutrimetabolomics: An Integrative Action for Metabolomic Analyses in Human Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, 1800384. [Google Scholar] [CrossRef]
- Wu, Y.; Li, L. Sample normalization methods in quantitative metabolomics. J. Chromatogr. A 2016, 1430, 80–95. [Google Scholar] [CrossRef]
- Emwas, A.H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar]
- Spicer, R.; Salek, R.M.; Moreno, P.; Cañueto, D.; Steinbeck, C. Navigating freely-available software tools for metabolomics analysis. Metabolomics 2017, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Castillo, S.; Gopalacharyulu, P.; Yetukuri, L.; Orešič, M. Algorithms and tools for the preprocessing of LC–MS metabolomics data. Chemom. Intell. Lab. Syst. 2011, 108, 23–32. [Google Scholar] [CrossRef]
- Sugimoto, M.; Kawakami, M.; Robert, M.; Soga, T.; Tomita, M. Bioinformatics Tools for Mass Spectroscopy-Based Metabolomic Data Processing and Analysis. Curr. Bioinform. 2012, 7, 96–108. [Google Scholar] [CrossRef]
- Katajamaa, M.; Orešič, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef]
- Hao, L.; Wang, J.; Page, D.; Asthana, S.; Zetterberg, H.; Carlsson, C.; Okonkwo, O.C.; Li, L. Comparative Evaluation of MS-based Metabolomics Software and Its Application to Preclinical Alzheimer’s Disease. Sci. Rep. 2018, 8, 9291. [Google Scholar] [CrossRef] [Green Version]
- Vettukattil, R. Preprocessing of Raw Metabonomic Data; Humana Press: New York, NY, USA, 2015; pp. 123–136. [Google Scholar]
- Vaclavik, L.; Lacina, O.; Hajslova, J.; Zweigenbaum, J. The use of high performance liquid chromatography-quadrupole time-of-flight mass spectrometry coupled to advanced data mining and chemometric tools for discrimination and classification of red wines according to their variety. Anal. Chim. Acta 2011, 685, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Sánchez De Medina, V.; Priego-Capote, F.; Luque De Castro, M.D. Characterization of refined edible oils enriched with phenolic extracts from olive leaves and pomace. J. Agric. Food Chem. 2012, 60, 5866–5873. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomoni, F.; Le Corguille, G.; Monsoor, M.; Landi, M.; Pericard, P.; Petera, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Lommen, A. MetAlign: Interface-Driven, Versatile Metabolomics Tool for Hyphenated Full-Scan Mass Spectrometry Data Preprocessing. Anal. Chem. 2009, 81, 3079–3086. [Google Scholar] [CrossRef]
- Bertsch, A.; Gröpl, C.; Reinert, K.; Kohlbacher, O. OpenMS and TOPP: Open Source Software for LC-MS Data Analysis. Methods Mol. Biol.. 2011, 696, 353–367. [Google Scholar]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Weber, R.J.M.; Lawson, T.N.; Salek, R.M.; Ebbels, T.M.D.; Glen, R.C.; Goodacre, R.; Griffin, J.L.; Haug, K.; Koulman, A.; Moreno, P.; et al. Computational tools and workflows in metabolomics: An international survey highlights the opportunity for harmonisation through Galaxy. Metabolomics 2017, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lu, Y.; Guo, Y.; Cao, H.; Wang, Q.; Shui, W. Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50–57. [Google Scholar] [CrossRef]
- Libiseller, G.; Dvorzak, M.; Kleb, U.; Gander, E.; Eisenberg, T.; Madeo, F.; Neumann, S.; Trausinger, G.; Sinner, F.; Pieber, T.; et al. IPO: A tool for automated optimization of XCMS parameters. BMC Bioinform. 2015, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Brunius, C.; Shi, L.; Landberg, R. Large-scale untargeted LC-MS metabolomics data correction using between-batch feature alignment and cluster-based within-batch signal intensity drift correction. Metabolomics 2016, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broeckling, C.D.; Afsar, F.A.; Neumann, S.; Ben-Hur, A.; Prenni, J.E. RAMClust: A novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.-C.; Burzykowski, T.; Berg, M. Evaluation of normalization methods to pave the way towards large-scale LC-MS-based metabolomics profiling experiments. OMICS 2013, 17, 473–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, H.; Ueda, K.; Kobayashi, Y.; Tsuyama, N.; Todoroki, K.; Min, J.Z.; Toyo’oka, T. The great importance of normalization of LC-MS data for highly-accurate non-targeted metabolomics. Biomed. Chromatogr. 2017, 31, 1–7. [Google Scholar] [CrossRef]
- Shen, X.; Gong, X.; Cai, Y.; Guo, Y.; Tu, J.; Li, H.; Zhang, T.; Wang, J.; Xue, F.; Zhu, Z.-J. Normalization and integration of large-scale metabolomics data using support vector regression. Metabolomics 2016, 12, 89. [Google Scholar] [CrossRef]
- Lee, J.; Park, J.; Lim, M.; Seong, S.J.; Seo, J.J.; Park, S.M.; Lee, H.W.; Yoon, Y.-R. Quantile normalization approach for liquid chromatography-mass spectrometry-based metabolomic data from healthy human volunteers. Anal. Sci. 2012, 28, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Nodzenski, M.; Muehlbauer, M.J.; Bain, J.R.; Reisetter, A.C.; Lowe, W.L.; Scholtens, D.M. Metabomxtr: An R package for mixture-model analysis of non-targeted metabolomics data. Bioinformatics 2014, 30, 3287–3288. [Google Scholar] [CrossRef] [Green Version]
- Chawade, A.; Alexandersson, E.; Levander, F. Normalyzer: A Tool for Rapid Evaluation of Normalization Methods for Omics Data Sets. J. Proteome Res. 2014, 13, 3114–3120. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, G.; Simpson, J.A.; Creek, D.J. NormalizeMets: Assessing, selecting and implementing statistical methods for normalizing metabolomics data. Metabolomics 2018, 14, 54. [Google Scholar] [CrossRef]
- Shi, L.; Westerhuis, J.A.; Rosén, J.; Landberg, R.; Brunius, C. Variable selection and validation in multivariate modelling. Bioinformatics 2018, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, I.; Liu, A. What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev. Neurother 2015, 15, 719–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, A.A.; Trygg, J.; Wuttge, D.M.; Sturfelt, G.; Theander, E.; Donten, M.; Moritz, T.; Sennbro, C.J.; Torell, F.; Lood, C.; et al. Metabolic profiling of systemic lupus erythematosus and comparison with primary Sjögren’s syndrome and systemic sclerosis. PLoS ONE 2016, 11, e0159384. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ochoa, Á.; Quirantes-Piné, R.; Borrás-Linares, I.; Gemperline, D.; Alarcón Riquelme, M.E.; Beretta, L.; Segura-Carretero, A. Urinary and plasma metabolite differences detected by HPLC-ESI-QTOF-MS in systemic sclerosis patients. J. Pharm. Biomed. Anal. 2019, 162, 82–90. [Google Scholar] [CrossRef]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0—Making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert; Humana Press: New York, NY, USA, 2017; pp. 339–368. [Google Scholar]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, F.; Hansen, B.; Karcher, W.; Sjöström, M.; Eriksson, L. Model validation by permutation tests: Applications to variable selection. J. Chemom. 1996, 10, 521–532. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis: Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil de la Fuente, A.; Grace Armitage, E.; Otero, A.; Barbas, C.; Godzien, J. Differentiating signals to make biological sense—A guide through databases for MS-based non-targeted metabolomics. Electrophoresis 2017, 38, 2242–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS-MUVR Models | nVar | Class (%) | AUC | nComp | p-Value |
|---|---|---|---|---|---|
| R Data | 15 | 86.8 | 0.931 | 2 | 1.38 × 10−6 |
| Profinder software Data | 67 | 81.7 | 0.893 | 3 | 2.80 × 10−5 |
| Profinder Software Methodology | R-Based Methodology | ||
|---|---|---|---|
 |  | | |
| Easy to use, user-friendly interface | License fee | Open source | Steep learning curve |
| High quality of the plots | Limited capacity to process a high number of samples | Greater number of packages, functions, and methods (e.g., normalization) | Low plot quality (plots obtained with the specific R packages used) |
| No need to transform the format of the data | Few normalization techniques. Difficulties to normalize large between-batch effects | High capacity for faster processing of a high number of samples | Data format transformation |
| Easy to inspect features, integration results, and MS spectra. Easy to predict molecular formula | Errors in peak integration | Possibility of carrying out all the steps of pre-processing and statistical analysis in the same environment | More cumbersome to show integration results, MS spectra, and to predict molecular formula |
| Easy to manually correct areas | Low control of the processing (only some parameters can be modified) | Flexibility and versatility | Some level of coding skills is required |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Ochoa, Á.; Quirantes-Piné, R.; Borrás-Linares, I.; Cádiz-Gurrea, M.d.l.L.; PRECISESADS Clinical Consortium; Alarcón Riquelme, M.E.; Brunius, C.; Segura-Carretero, A. A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites 2020, 10, 28. https://doi.org/10.3390/metabo10010028
Fernández-Ochoa Á, Quirantes-Piné R, Borrás-Linares I, Cádiz-Gurrea MdlL, PRECISESADS Clinical Consortium, Alarcón Riquelme ME, Brunius C, Segura-Carretero A. A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites. 2020; 10(1):28. https://doi.org/10.3390/metabo10010028
Chicago/Turabian StyleFernández-Ochoa, Álvaro, Rosa Quirantes-Piné, Isabel Borrás-Linares, María de la Luz Cádiz-Gurrea, PRECISESADS Clinical Consortium, Marta E. Alarcón Riquelme, Carl Brunius, and Antonio Segura-Carretero. 2020. "A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics" Metabolites 10, no. 1: 28. https://doi.org/10.3390/metabo10010028
APA StyleFernández-Ochoa, Á., Quirantes-Piné, R., Borrás-Linares, I., Cádiz-Gurrea, M. d. l. L., PRECISESADS Clinical Consortium, Alarcón Riquelme, M. E., Brunius, C., & Segura-Carretero, A. (2020). A Case Report of Switching from Specific Vendor-Based to R-Based Pipelines for Untargeted LC-MS Metabolomics. Metabolites, 10(1), 28. https://doi.org/10.3390/metabo10010028