Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis

 ,
, 
Abstract
:1. Introduction
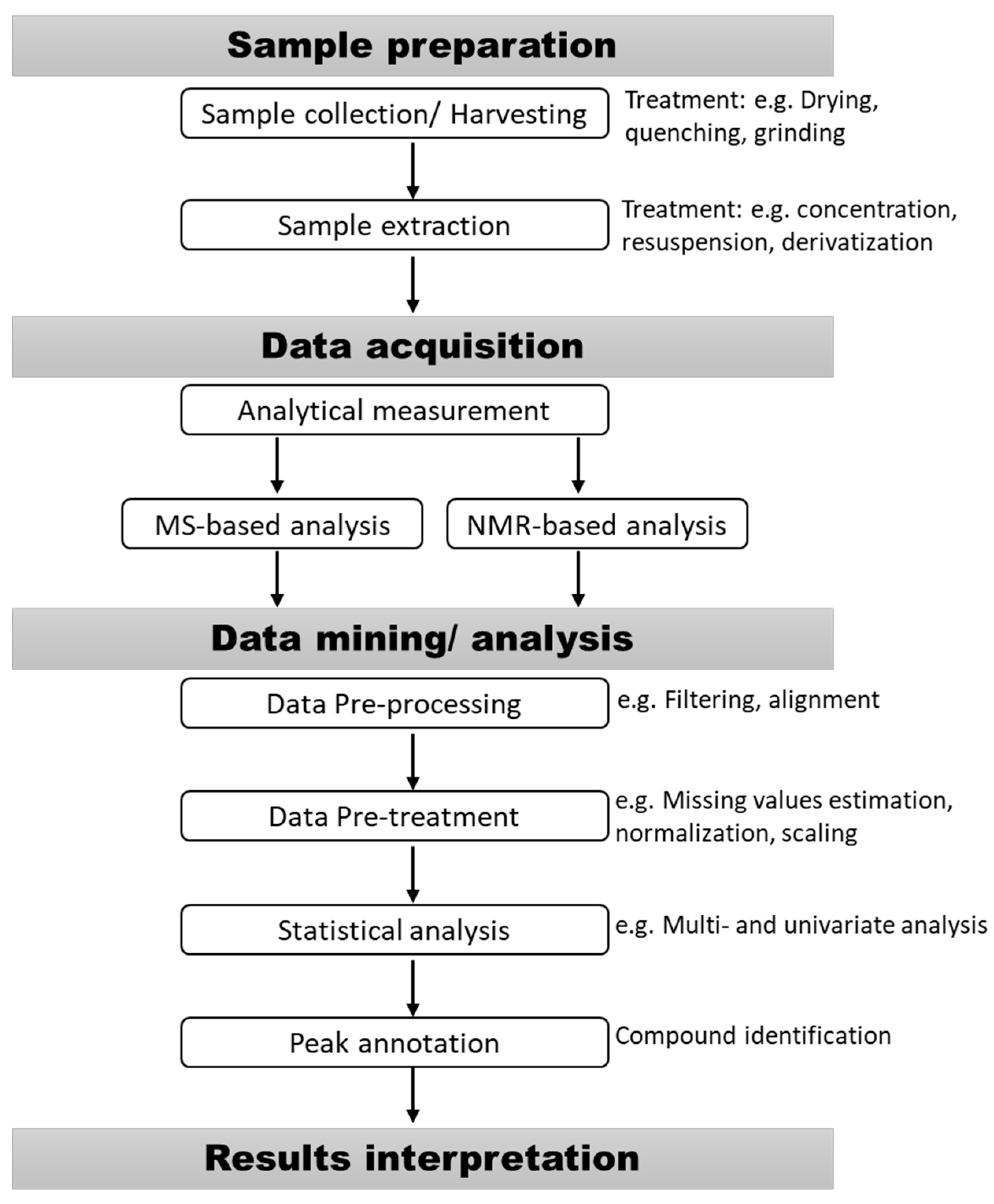
2. Sample Preparation for Metabolomic Studies
2.1. Sample Collection
2.2. Harvesting Methods
2.3. Sample Extraction
2.4. Extract Concentration, Dilution, Enrichment, and Re-Suspension
3. Analytical Methods for Metabolites Analysis
3.1. Gas Chromatography-Mass Spectrometry (GC-MS)
3.2. Liquid Chromatography-Mass Spectrometry (LC-MS)
3.3. Capillary Electrophoresis-Mass Spectrometry (CE-MS)
3.4. Nuclear Magnetic Resonance (NMR)
3.4.1. 2D NMR Based Metabolomics Strategies
3.4.2. Solid State NMR Based Metabolomics
3.4.3. Hyphenated NMR-Based Metabolomics
4. Metabolomic Data Processing and Interpretation
5. Successful Applications of Metabolomics in Natural Products Discovery
5.1. Metabolomics for Secondary Metabolites Identification
5.2. Metabolomics in Defining or Refining the Pathway Structure of Plant Natural Products
5.3. Metabolomics to Aid Metabolic Engineering of Secondary Metabolites Production
5.4. Metabolomics and Dereplication
5.5. Metabolomics for Quality Control of Natural Products
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural Product Drug Discovery in the Next Millennium. Pharm. Biol. 2001, 39, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Scossa, F.; Benina, M.; Alseekh, S.; Zhang, Y.; Fernie, A.R. The Integration of Metabolomics and Next-Generation Sequencing Data to Elucidate the Pathways of Natural Product Metabolism in Medicinal Plants. Planta Med. 2018, 84, 855–873. [Google Scholar] [CrossRef] [Green Version]
- Skirycz, A.; Kierszniowska, S.; Méret, M.; Willmitzer, L.; Tzotzos, G. Medicinal Bioprospecting of the Amazon Rainforest: A Modern Eldorado? Trends Biotechnol. 2016, 34, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Nagatomi, Y.; Harayama, K.; Bamba, T. Development of an analytical method for polycyclic aromatic hydrocarbons in coffee beverages and dark beer using novel high-sensitivity technique of supercritical fluid chromatography/mass spectrometry. J. Biosci. Bioeng. 2018, 126, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.; Piris, A.J.; Ruiz-Rodriguez, A.; Prodanov, M.; Soler-Rivas, C. Extraction of bioactive compounds against cardiovascular diseases from Lentinula edodes using a sequential extraction method. Biotechnol. Prog. 2018, 34, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Joana Gil-Chávez, G.; Villa, J.A.; Fernando Ayala-Zavala, J.; Basilio Heredia, J.; Sepulveda, D.; Yahia, E.M.; González-Aguilar, G.A. Technologies for Extraction and Production of Bioactive Compounds to be Used as Nutraceuticals and Food Ingredients: An Overview. Compr. Rev. Food Sci. Food Saf. 2013, 12, 5–23. [Google Scholar] [CrossRef]
- Gan, Z.; Liang, Z.; Chen, X.; Wen, X.; Wang, Y.; Li, M.; Ni, Y. Separation and preparation of 6-gingerol from molecular distillation residue of Yunnan ginger rhizomes by high-speed counter-current chromatography and the antioxidant activity of ginger oils in vitro. J. Chromatogr. B 2016, 1011, 99–107. [Google Scholar] [CrossRef]
- Williams, S.R.; Oatley, D.L.; Abdrahman, A.; Butt, T.; Nash, R. Membrane Technology for the Improved Separation of Bioactive Compounds. Procedia Eng. 2012, 44, 2112–2114. [Google Scholar] [CrossRef] [Green Version]
- Salam, A.M.; Lyles, J.T.; Quave, C.L. Methods in the Extraction and Chemical Analysis of Medicinal Plants. In Methods and Techniques in Ethnobiology and Ethnoecology; Albuquerque, U.P., de Lucena, R.F.P., Cruz da Cunha, L.V.F., Alves, R.R.N., Eds.; Springer: New York, NY, USA, 2019; pp. 257–283. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinette, S.L.; Brüschweiler, R.; Schroeder, F.C.; Edison, A.S. NMR in metabolomics and natural products research: Two sides of the same coin. Acc. Chem. Res. 2012, 45, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Tyagi, A.K.; Singh, R.P.; Chan, D.C.F.; Agarwal, R. Synergistic Anti-Cancer Effects of Grape Seed Extract and Conventional Cytotoxic Agent Doxorubicin Against Human Breast Carcinoma Cells. Breast Cancer Res. Treat. 2004, 85, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chusri, S.; Siriyong, T.; Na-Phatthalung, P.; Voravuthikunchai, S.P. Synergistic effects of ethnomedicinal plants of Apocynaceae family and antibiotics against clinical isolates of Acinetobacter baumannii. Asian Pac. J. Trop. Med. 2014, 7, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Skroza, D.; Generalić Mekinić, I.; Svilović, S.; Šimat, V.; Katalinić, V. Investigation of the potential synergistic effect of resveratrol with other phenolic compounds: A case of binary phenolic mixtures. J. Food Compos. Anal. 2015, 38, 13–18. [Google Scholar] [CrossRef]
- Shyur, L.-F.; Yang, N.-S. Metabolomics for phytomedicine research and drug development. Curr. Opin. Chem. Biol. 2008, 12, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lamers, R.J.; Korthout, H.A.; van Nesselrooij, J.H.; Witkamp, R.F.; van der Heijden, R.; Voshol, P.J.; Havekes, L.M.; Verpoorte, R.; van der Greef, J. Metabolomics in the context of systems biology: Bridging traditional Chinese medicine and molecular pharmacology. Phytother. Res. 2005, 19, 173–182. [Google Scholar] [CrossRef]
- Ulrich-Merzenich, G.; Zeitler, H.; Jobst, D.; Panek, D.; Vetter, H.; Wagner, H. Application of the “-Omic-” technologies in phytomedicine. Phytomedicine 2007, 14, 70–82. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Bekri, S. Advances in metabolome information retrieval: Turning chemistry into biology. Part I: Analytical chemistry of the metabolome. J. Inherit. Metab. Dis. 2018, 41, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Alseekh, S.; Fernie, A.R. Metabolomics 20 years on: What have we learned and what hurdles remain? Plant J. 2018, 94, 933–942. [Google Scholar] [CrossRef]
- Pinto, R.C. Chemometrics Methods and Strategies in Metabolomics. In Metabolomics: From Fundamentals to Clinical Applications; Sussulini, A., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 163–190. [Google Scholar] [CrossRef]
- Farrag, A.R.H.; Abdallah, H.M.I.; Khattab, A.R.; Elshamy, A.I.; Gendy, A.E.-N.G.E.; Mohamed, T.A.; Farag, M.A.; Efferth, T.; Hegazy, M.-E.F. Antiulcer activity of Cyperus alternifolius in relation to its UPLC-MS metabolite fingerprint: A mechanistic study. Phytomedicine 2019, 62, 152970. [Google Scholar] [CrossRef]
- Yang, S.Y.; Kim, H.K.; Lefeber, A.W.M.; Erkelens, C.; Angelova, N.; Choi, Y.H.; Verpoorte, R. Application of Two-Dimensional Nuclear Magnetic Resonance Spectroscopy to Quality Control of Ginseng Commercial Products. Planta Med. 2006, 72, 364–369. [Google Scholar] [CrossRef]
- Zeng, Z.-D.; Liang, Y.-Z.; Chau, F.-T.; Chen, S.; Daniel, M.K.-W.; Chan, C.-O. Mass spectral profiling: An effective tool for quality control of herbal medicines. Anal. Chim. Acta 2007, 604, 89–98. [Google Scholar] [CrossRef]
- Dunn, W.B.; Erban, A.; Weber, R.J.M.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J.; et al. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- Perez de Souza, L.; Naake, T.; Tohge, T.; Fernie, A.R. From chromatogram to analyte to metabolite. How to pick horses for courses from the massive web resources for mass spectral plant metabolomics. GigaScience 2017, 6, gix037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef] [Green Version]
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the t-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef]
- Riekeberg, E.; Powers, R. New frontiers in metabolomics: From measurement to insight. F1000Research 2017, 6, 1148. [Google Scholar] [CrossRef] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Blainey, P.; Krzywinski, M.; Altman, N. Replication. Nat. Methods 2014, 11, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Verpoorte, R. Sample preparation for plant metabolomics. Phytochem. Anal. 2010, 21, 4–13. [Google Scholar] [CrossRef]
- Ernst, M.; Silva, D.B.; Silva, R.R.; Vencio, R.Z.; Lopes, N.P. Mass spectrometry in plant metabolomics strategies: From analytical platforms to data acquisition and processing. Nat. Prod. Rep. 2014, 31, 784–806. [Google Scholar] [CrossRef]
- Fernie, A.R.; Trethewey, R.N.; Krotzky, A.J.; Willmitzer, L. Innovation—Metabolite profiling: From diagnostics to systems biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 763–769. [Google Scholar] [CrossRef]
- Dunn, W.B.; Winder, C.L. Sample Preparation Related to the Intracellular Metabolome of Yeast: Methods for Quenching, Extraction, and Metabolite Quantitation. Method Enzym. 2011, 500, 277–297. [Google Scholar] [CrossRef]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS. Analyst 2014, 139, 2265–2276. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for Metabolomics: Access to the Metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef]
- Salem, M.A.; Giavalisco, P. Semi-targeted Lipidomics of Plant Acyl Lipids Using UPLC-HR-MS in Combination with a Data-Independent Acquisition Mode. In Plant Metabolomics: Methods and Protocols, Methods in Molecular Biology; António, C., Ed.; Springer: New York, NY, USA; Humana Press: Totowa, NJ, USA, 2018; Volume 1778, pp. 137–155. [Google Scholar]
- Salem, M.; Bernach, M.; Bajdzienko, K.; Giavalisco, P. A Simple Fractionated Extraction Method for the Comprehensive Analysis of Metabolites, Lipids, and Proteins from a Single Sample. J. Vis. Exp. 2017, 124, e55802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raterink, R.-J.; Lindenburg, P.W.; Vreeken, R.J.; Ramautar, R.; Hankemeier, T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. TrAC Trends Anal. Chem. 2014, 61, 157–167. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.H.S. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Weckwerth, W.; Wenzel, K.; Fiehn, O. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 2004, 4, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kopka, J.; Trethewey, R.N.; Willmitzer, L. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal. Chem. 2000, 72, 3573–3580. [Google Scholar] [CrossRef]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Giavalisco, P.; Li, Y.; Matthes, A.; Eckhardt, A.; Hubberten, H.M.; Hesse, H.; Segu, S.; Hummel, J.; Kohl, K.; Willmitzer, L. Elemental formula annotation of polar and lipophilic metabolites using 13C, 15N and 34S isotope labelling, in combination with high-resolution mass spectrometry. Plant J. Cell Mol. Biol. 2011, 68, 364–376. [Google Scholar] [CrossRef]
- Bozek, K.; Wei, Y.; Yan, Z.; Liu, X.; Xiong, J.; Sugimoto, M.; Tomita, M.; Paabo, S.; Sherwood, C.C.; Hof, P.R.; et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron 2015, 85, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Khrameeva, E.E.; Bozek, K.; He, L.; Yan, Z.; Jiang, X.; Wei, Y.; Tang, K.; Gelfand, M.S.; Prufer, K.; Kelso, J.; et al. Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nat. Commun. 2014, 5, 3584. [Google Scholar] [CrossRef] [Green Version]
- Bozek, K.; Wei, Y.; Yan, Z.; Liu, X.; Xiong, J.; Sugimoto, M.; Tomita, M.; Paabo, S.; Pieszek, R.; Sherwood, C.C.; et al. Exceptional evolutionary divergence of human muscle and brain metabolomes parallels human cognitive and physical uniqueness. PLoS Biol. 2014, 12, e1001871. [Google Scholar] [CrossRef] [Green Version]
- Kuyukina, M.S.; Ivshina, I.B.; Philp, J.C.; Christofi, N.; Dunbar, S.A.; Ritchkova, M.I. Recovery of Rhodococcus biosurfactants using methyl tertiary-butyl ether extraction. J. Microbiol. Methods 2001, 46, 149–156. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.A.; Li, Y.; Bajdzienko, K.; Fisahn, J.; Watanabe, M.; Hoefgen, R.; Schottler, M.A.; Giavalisco, P. RAPTOR Controls Developmental Growth Transitions by Altering the Hormonal and Metabolic Balance. Plant Physiol. 2018, 177, 565–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, M.A.; Meyer, A.; Ali, S.E.; Salem, M.A.; Giavalisco, P.; Westphal, H.; Wessjohann, L.A. Comparative Metabolomics Approach Detects Stress-Specific Responses during Coral Bleaching in Soft Corals. J. Proteome Res. 2018, 17, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Alhajturki, D.; Muralidharan, S.; Nurmi, M.; Rowan, B.A.; Lunn, J.E.; Boldt, H.; Salem, M.A.; Alseekh, S.; Jorzig, C.; Feil, R.; et al. Dose-dependent interactions between two loci trigger altered shoot growth in BG-5 x Krotzenburg-0 (Kro-0) hybrids of Arabidopsis thaliana. New Phytol. 2018, 217, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.A.; Li, Y.; Wiszniewski, A.; Giavalisco, P. Regulatory-associated protein of TOR (RAPTOR) alters the hormonal and metabolic composition of Arabidopsis seeds, controlling seed morphology, viability and germination potential. Plant J. Cell Mol. Biol. 2017, 92, 525–545. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.A.; Juppner, J.; Bajdzienko, K.; Giavalisco, P. Protocol: A fast, comprehensive and reproducible one-step extraction method for the rapid preparation of polar and semi-polar metabolites, lipids, proteins, starch and cell wall polymers from a single sample. Plant Methods 2016, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Doppler, M.; Kluger, B.; Bueschl, C.; Schneider, C.; Krska, R.; Delcambre, S.; Hiller, K.; Lemmens, M.; Schuhmacher, R. Stable Isotope-Assisted Evaluation of Different Extraction Solvents for Untargeted Metabolomics of Plants. Int. J. Mol. Sci. 2016, 17, 1017. [Google Scholar] [CrossRef] [Green Version]
- Roessner, U.; Dias, D.A. Plant Tissue Extraction for Metabolomics. Metab. Tools Nat. Prod. Discov. Methods Protoc. 2013, 1055, 21–28. [Google Scholar] [CrossRef]
- De Vos, R.C.; Moco, S.; Lommen, A.; Keurentjes, J.J.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal. Chem. TrAC 2014, 61, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernie, A.R.; Aharoni, A.; Willmitzer, L.; Stitt, M.; Tohge, T.; Kopka, J.; Carroll, A.J.; Saito, K.; Fraser, P.D.; DeLuca, V. Recommendations for reporting metabolite data. Plant Cell 2011, 23, 2477–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.A.; Strack, D. Phytochemistry meets genome analysis, and beyond. Phytochemistry 2003, 62, 815–816. [Google Scholar] [CrossRef] [Green Version]
- Rai, A.; Saito, K.; Yamazaki, M. Integrated omics analysis of specialized metabolism in medicinal plants. Plant J. Cell Mol. Biol. 2017, 90, 764–787. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; Troncoso-Ponce, M.A.; Ratcliffe, R.G. 1H NMR metabolite fingerprinting and metabolomic analysis of perchloric acid extracts from plant tissues. Nat. Protoc. 2008, 3, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Fernie, A.R. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protoc. 2010, 5, 1210–1227. [Google Scholar] [CrossRef]
- Sumner, L.W.; Mendes, P.; Dixon, R.A. Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62, 817–836. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomic—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Kopka, J.; Fernie, A.; Weckwerth, W.; Gibon, Y.; Stitt, M. Metabolite profiling in plant biology: Platforms and destinations. Genome Biol. 2004, 5, 109. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [Green Version]
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 2001, 13, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Halket, J.M.; Przyborowska, A.; Stein, S.E.; Mallard, W.G.; Down, S.; Chalmers, R.A. Deconvolution gas chromatography/mass spectrometry of urinary organic acids-potential for pattern recognition and automated identification of metabolic disorders. Rapid Commun. Mass Spectrom. 1999, 13, 279–284. [Google Scholar] [CrossRef]
- Saito, K.; Matsuda, F. Metabolomics for Functional Genomics, Systems Biology, and Biotechnology. Annu. Rev. Plant Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dormann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauer, N.; Steinhauser, D.; Strelkov, S.; Schomburg, D.; Allison, G.; Moritz, T.; Lundgren, K.; Roessner-Tunali, U.; Forbes, M.G.; Willmitzer, L.; et al. GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett. 2005, 579, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Lange, B.M. Open-access metabolomics databases for natural product research: Present capabilities and future potential. Front. Bioeng. Biotechnol. 2015, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sirimanne, S.R.; Patterson, D.G., Jr.; Needham, L.L. Comprehensive two-dimensional gas chromatography for the fast separation and determination of pesticides extracted from human serum. Anal. Chem. 1994, 66, 3086–3092. [Google Scholar] [CrossRef]
- Mondello, L.; Casillia, A.; Tranchida, P.Q.; Dugo, G.; Dugo, P. Comprehensive two-dimensional gas chromatography in combination with rapid scanning quadrupole mass spectrometry in perfume analysis. J. Chromatogr. A 2005, 1067, 235–243. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, H.; Reim, A.; Charles, P.D.; Northage, A.; Jackson, D.; Parry, I.; Kessler, B.M. Optimizing 2D gas chromatography mass spectrometry for robust tissue, serum and urine metabolite profiling. Talanta 2017, 165, 685–691. [Google Scholar] [CrossRef]
- Egert, B.; Weinert, C.H.; Kulling, S.E. A peaklet-based generic strategy for the untargeted analysis of comprehensive two-dimensional gas chromatography mass spectrometry data sets. J. Chromatogr. A 2015, 1405, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Hartmann, C.; J Marriott, P. Multidimensional gas chromatography methods for bioanalytical research. Bioanalysis 2014, 6, 2461–2479. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Edwards, M.; Gorecki, T. Optimization aspects of comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2012, 1255, 38–55. [Google Scholar] [CrossRef]
- Hill, C.B.; Roessner, U. Metabolic Profiling of Plants by GC–MS. In The Handbook of Plant Metabolomics; Weckwerth, W., Kahl, G., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1–23. [Google Scholar] [CrossRef]
- Iijima, Y.; Nakamura, Y.; Ogata, Y.; Tanaka, K.; Sakurai, N.; Suda, K.; Suzuki, T.; Suzuki, H.; Okazaki, K.; Kitayama, M.; et al. Metabolite annotations based on the integration of mass spectral information. Plant J. Cell Mol. Biol. 2008, 54, 949–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, F.; Hirai, M.Y.; Sasaki, E.; Akiyama, K.; Yonekura-Sakakibara, K.; Provart, N.J.; Sakurai, T.; Shimada, Y.; Saito, K. AtMetExpress development: A phytochemical atlas of Arabidopsis development. Plant Physiol. 2010, 152, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, F.; Yonekura-Sakakibara, K.; Niida, R.; Kuromori, T.; Shinozaki, K.; Saito, K. MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J. Cell Mol. Biol. 2009, 57, 555–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, Y.; Shimojima, M.; Sawada, Y.; Toyooka, K.; Narisawa, T.; Mochida, K.; Tanaka, H.; Matsuda, F.; Hirai, A.; Hirai, M.Y.; et al. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell 2009, 21, 892–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, M.A.; Giavalisco, P. Semi-targeted Lipidomics of Plant Acyl Lipids Using UPLC-HR-MS in Combination with a Data-Independent Acquisition Mode. Methods Mol. Biol. 2018, 1778, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Jones, A.D.; Last, R.L. LC-MS/MS assay for protein amino acids and metabolically related compounds for large-scale screening of metabolic phenotypes. Anal. Chem. 2007, 79, 8067–8075. [Google Scholar] [CrossRef]
- Bocker, S.; Rasche, F. Towards de novo identification of metabolites by analyzing tandem mass spectra. Bioinformatics 2008, 24, i49–i55. [Google Scholar] [CrossRef]
- Keurentjes, J.J.; Fu, J.; de Vos, C.H.; Lommen, A.; Hall, R.D.; Bino, R.J.; van der Plas, L.H.; Jansen, R.C.; Vreugdenhil, D.; Koornneef, M. The genetics of plant metabolism. Nat. Genet. 2006, 38, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giavalisco, P.; Kohl, K.; Hummel, J.; Seiwert, B.; Willmitzer, L. 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 2009, 81, 6546–6551. [Google Scholar] [CrossRef] [PubMed]
- Shahaf, N.; Rogachev, I.; Heinig, U.; Meir, S.; Malitsky, S.; Battat, M.; Wyner, H.; Zheng, S.; Wehrens, R.; Aharoni, A. The WEIZMASS spectral library for high-confidence metabolite identification. Nat. Commun. 2016, 7, 12423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giavalisco, P.; Hummel, J.; Lisec, J.; Inostroza, A.C.; Catchpole, G.; Willmitzer, L. High-resolution direct infusion-based mass spectrometry in combination with whole 13C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Anal. Chem. 2008, 80, 9417–9425. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Kusano, M.; Kobayashi, M.; Tohge, T.; Yonekura-Sakakibara, K.; Kogure, N.; Yamazaki, M.; Kitajima, M.; Saito, K.; Takayama, H. Metabolomics-oriented isolation and structure elucidation of 37 compounds including two anthocyanins from Arabidopsis thaliana. Phytochemistry 2009, 70, 1017–1029. [Google Scholar] [CrossRef]
- Tohge, T.; Fernie, A.R. Web-based resources for mass-spectrometry-based metabolomics: A user’s guide. Phytochemistry 2009, 70, 450–456. [Google Scholar] [CrossRef]
- Ishii, N.; Nakahigashi, K.; Baba, T.; Robert, M.; Soga, T.; Kanai, A.; Hirasawa, T.; Naba, M.; Hirai, K.; Hoque, A.; et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 2007, 316, 593–597. [Google Scholar] [CrossRef]
- Fernie, A.R.; Tohge, T. The Genetics of Plant Metabolism. Annu. Rev. Genet. 2017, 51, 287–310. [Google Scholar] [CrossRef]
- Williams, B.J.; Cameron, C.J.; Workman, R.; Broeckling, C.D.; Sumner, L.W.; Smith, J.T. Amino acid profiling in plant cell cultures: An inter-laboratory comparison of CE-MS and GC-MS. Electrophoresis 2007, 28, 1371–1379. [Google Scholar] [CrossRef]
- Soga, T.; Imaizumi, M. Capillary electrophoresis method for the analysis of inorganic anions, organic acids, amino acids, nucleotides, carbohydrates and other anionic compounds. Electrophoresis 2001, 22, 3418–3425. [Google Scholar] [CrossRef]
- Ren, J.-L.; Zhang, A.-H.; Kong, L.; Wang, X.-J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv. 2018, 8, 22335–22350. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based plant metabolomics: Where do we stand, where do we go? Trends Biotechnol. 2011, 29, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Saifullah; Khan, S.; Wilson, E.G.; Kricun, S.D.P.; Meissner, A.; Goraler, S.; Deelder, A.M.; Choi, Y.H.; Verpoorte, R. Metabolic classification of South American Ilex species by NMR-based metabolomics. Phytochemistry 2010, 71, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Mahrous, E.A.; Farag, M.A. Two dimensional NMR spectroscopic approaches for exploring plant metabolome: A review. J. Adv. Res. 2015, 6, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grkovic, T.; Pouwer, R.H.; Vial, M.-L.; Gambini, L.; Noël, A.; Hooper, J.N.A.; Wood, S.A.; Mellick, G.D.; Quinn, R.J. NMR Fingerprints of the Drug-like Natural-Product Space Identify Iotrochotazine A: A Chemical Probe to Study Parkinson’s Disease. Angew. Chem. Int. Ed. 2014, 53, 6070–6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starks, C.M.; Williams, R.B.; Norman, V.L.; Lawrence, J.A.; O’Neil-Johnson, M.; Eldridge, G.R. Phenylpropanoids from Phragmipedium calurum and their antiproliferative activity. Phytochemistry 2012, 82, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.-S.; Short, L.C.; Syage, J.A.; Potvin, M.; Curtis, J.M. Liquid chromatography–atmospheric pressure photoionization-mass spectrometry analysis of triacylglycerol lipids—Effects of mobile phases on sensitivity. J. Chromatogr. A 2007, 1173, 88–97. [Google Scholar] [CrossRef]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc. 2010, 5, 536. [Google Scholar] [CrossRef]
- Tawfike, A.F.; Viegelmann, C.; Edrada-Ebel, R. Metabolomics and Dereplication Strategies in Natural Products. In Metabolomics Tools for Natural Product Discovery: Methods and Protocols; Roessner, U., Dias, D.A., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 227–244. [Google Scholar] [CrossRef]
- Zhang, L.; Hatzakis, E.; Patterson, A.D. NMR-Based Metabolomics and Its Application in Drug Metabolism and Cancer Research. Curr. Pharmacol. Rep. 2016, 2, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Simmler, C.; Napolitano, J.G.; McAlpine, J.B.; Chen, S.-N.; Pauli, G.F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. [Google Scholar] [CrossRef]
- Nagana Gowda, G.A.; Raftery, D. Can NMR solve some significant challenges in metabolomics? J. Magn. Reson. 2015, 260, 144–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halabalaki, M.; Vougogiannopoulou, K.; Mikros, E.; Skaltsounis, A.L. Recent advances and new strategies in the NMR-based identification of natural products. Curr. Opin. Biotechnol. 2014, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, H.; Moskau, D.; Spraul, M. Cryogenically cooled probes—A leap in NMR technology. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 131–155. [Google Scholar] [CrossRef]
- Dona, A.C. CHAPTER 1 Instrumental Platforms for NMR-based Metabolomics. In NMR-Based Metabolomics; The Royal Society of Chemistry: London, UK, 2018; pp. 1–21. [Google Scholar]
- Deborde, C.; Moing, A.; Roch, L.; Jacob, D.; Rolin, D.; Giraudeau, P. Plant metabolism as studied by NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 61–97. [Google Scholar] [CrossRef] [PubMed]
- Anklin, C. Chapter 3 Small-volume NMR: Microprobes and Cryoprobes. In Modern NMR Approaches to the Structure Elucidation of Natural Products: Volume 1: Instrumentation and Software; The Royal Society of Chemistry: London, UK, 2016; Volume 1, pp. 38–57. [Google Scholar]
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forseth, R.R.; Schroeder, F.C. NMR-spectroscopic analysis of mixtures: From structure to function. Curr. Opin. Chem. Biol. 2011, 15, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa-Carballal, R.; Fernandez-Megia, E.; Jimenez, C.; Riguera, R. NMR methods for unravelling the spectra of complex mixtures. Nat. Prod. Rep. 2011, 28, 78–98. [Google Scholar] [CrossRef]
- Rabenstein, D.L.; Fan, S. Proton nuclear magnetic resonance spectroscopy of aqueous solutions: Complete elimination of the water resonance by spin-spin relaxation. Anal. Chem. 1986, 58, 3178–3184. [Google Scholar] [CrossRef]
- Johnson, C.S. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Balayssac, S.; Trefi, S.; Gilard, V.; Malet-Martino, M.; Martino, R.; Delsuc, M.-A. 2D and 3D DOSY 1H NMR, a useful tool for analysis of complex mixtures: Application to herbal drugs or dietary supplements for erectile dysfunction. J. Pharm. Biomed. Anal. 2009, 50, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Politi, M.; Zloh, M.; Pintado, M.E.; Castro, P.M.L.; Heinrich, M.; Prieto, J.M. Direct metabolic fingerprinting of commercial herbal tinctures by nuclear magnetic resonance spectroscopy and mass spectrometry. Phytochem. Anal. 2009, 20, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Yasuda, T.; Fukushi, E.; Kawabata, J.; Sekiguchi, M.; Fromont, J.; Kobayashi, J. Agesamides A and B, Bromopyrrole Alkaloids from Sponge Agelas Species: Application of DOSY for Chemical Screening of New Metabolites. Org. Lett. 2006, 8, 4235–4238. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Viant, M.R. Two-dimensional J-resolved NMR spectroscopy: Review of a key methodology in the metabolomics toolbox. Phytochem. Anal. 2010, 21, 22–32. [Google Scholar] [CrossRef]
- Georgiev, M.I.; Ali, K.; Alipieva, K.; Verpoorte, R.; Choi, Y.H. Metabolic differentiations and classification of Verbascum species by NMR-based metabolomics. Phytochemistry 2011, 72, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.A.; Faulkner, S.; Nilsson, M.; Morris, G.A. Pure Shift 1H NMR: A Resolution of the Resolution Problem? Angew. Chem. 2010, 122, 3993–3995. [Google Scholar] [CrossRef]
- Lopez, J.M.; Cabrera, R.; Maruenda, H. Ultra-Clean Pure Shift 1H-NMR applied to metabolomics profiling. Sci. Rep. 2019, 9, 6900. [Google Scholar] [CrossRef]
- Breton, R.C.; Reynolds, W.F. Using NMR to identify and characterize natural products. Nat. Prod. Rep. 2013, 30, 501–524. [Google Scholar] [CrossRef]
- Parkinson, J.A. Chapter 2—NMR Spectroscopy Methods in Metabolic Phenotyping. In The Handbook of Metabolic Phenotyping; Lindon, J.C., Nicholson, J.K., Holmes, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 53–96. [Google Scholar] [CrossRef]
- Koskela, H.; Heikkilä, O.; Kilpeläinen, I.; Heikkinen, S. Quantitative two-dimensional HSQC experiment for high magnetic field NMR spectrometers. J. Magn. Reson. 2010, 202, 24–33. [Google Scholar] [CrossRef]
- Dossey, A.T.; Walse, S.S.; Conle, O.V.; Edison, A.S. Parectadial, a Monoterpenoid from the Defensive Spray of Parectatosoma mocquerysi. J. Nat. Prod. 2007, 70, 1335–1338. [Google Scholar] [CrossRef]
- Farag, M.A.; Porzel, A.; Al-Hammady, M.A.; Hegazy, M.-E.F.; Meyer, A.; Mohamed, T.A.; Westphal, H.; Wessjohann, L.A. Soft Corals Biodiversity in the Egyptian Red Sea: A Comparative MS and NMR Metabolomics Approach of Wild and Aquarium Grown Species. J. Proteome Res. 2016, 15, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Farag, M.A.; Mahrous, E.A.; Lübken, T.; Porzel, A.; Wessjohann, L. Classification of commercial cultivars of Humulus lupulus L. (hop) by chemometric pixel analysis of two dimensional nuclear magnetic resonance spectra. Metabolomics 2014, 10, 21–32. [Google Scholar] [CrossRef]
- Schroeder, F.C.; Gibson, D.M.; Churchill, A.C.L.; Sojikul, P.; Wursthorn, E.J.; Krasnoff, S.B.; Clardy, J. Differential Analysis of 2D NMR Spectra: New Natural Products from a Pilot-Scale Fungal Extract Library. Angew. Chem. Int. Ed. 2007, 46, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Pungaliya, C.; Srinivasan, J.; Fox, B.W.; Malik, R.U.; Ludewig, A.H.; Sternberg, P.W.; Schroeder, F.C. A shortcut to identifying small molecule signals that regulate behavior and development in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2009, 106, 7708–7713. [Google Scholar] [CrossRef] [Green Version]
- Butcher, R.A.; Schroeder, F.C.; Fischbach, M.A.; Straight, P.D.; Kolter, R.; Walsh, C.T.; Clardy, J. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 1506–1509. [Google Scholar] [CrossRef] [Green Version]
- McCarney, E.R.; Dykstra, R.; Galvosas, P. Evaluation of benchtop NMR Diffusion Ordered Spectroscopy for small molecule mixture analysis. Magn. Reson. Imaging 2019, 56, 103–109. [Google Scholar] [CrossRef]
- Blümich, B. Low-field and benchtop NMR. J. Magn. Reson. 2019, 306, 27–35. [Google Scholar] [CrossRef]
- Gouilleux, B.; Marchand, J.; Charrier, B.; Remaud, G.S.; Giraudeau, P. High-throughput authentication of edible oils with benchtop Ultrafast 2D NMR. Food Chem. 2018, 244, 153–158. [Google Scholar] [CrossRef]
- Kruk, J.; Doskocz, M.; Jodłowska, E.; Zacharzewska, A.; Łakomiec, J.; Czaja, K.; Kujawski, J. NMR Techniques in Metabolomic Studies: A Quick Overview on Examples of Utilization. Appl. Magn. Reson. 2017, 48, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, E.M.S.; Iglesias, M.J.; Ortiz, F.L.; Pérez, I.S.; Galera, M.M. Study of the suitability of HRMAS NMR for metabolic profiling of tomatoes: Application to tissue differentiation and fruit ripening. Food Chem. 2010, 122, 877–887. [Google Scholar] [CrossRef]
- Daolio, C.; Beltrame, F.L.; Ferreira, A.G.; Cass, Q.B.; Cortez, D.A.G.; Ferreira, M.M.C. Classification of commercial Catuaba samples by NMR, HPLC and chemometrics. Phytochem. Anal. 2008, 19, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Broberg, A.; Kenne, L. Use of High-Resolution Magic Angle Spinning Nuclear Magnetic Resonance Spectroscopy for in Situ Studies of Low-Molecular-Mass Compounds in Red Algae. Anal. Biochem. 2000, 284, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Miglietta, M.L.; Lamanna, R. 1H HR-MAS NMR of carotenoids in aqueous samples and raw vegetables. Magn. Reson. Chem. 2006, 44, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Sekiyama, Y.; Chikayama, E.; Kikuchi, J. Profiling Polar and Semipolar Plant Metabolites throughout Extraction Processes Using a Combined Solution-State and High-Resolution Magic Angle Spinning NMR Approach. Anal. Chem. 2010, 82, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, M.; Heerschap, A. Effect of Cation Binding on the Proton Chemical Shifts and the Spin–Spin Coupling Constant of Citrate. J. Magn. Reson. Ser. B 1996, 112, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Jaroszewski, J.W. Hyphenated NMR Methods in Natural Products Research, Part 1: Direct Hyphenation. Planta Med. 2005, 71, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Seger, C.; Sturm, S.; Stuppner, H. Mass spectrometry and NMR spectroscopy: Modern high-end detectors for high resolution separation techniques—State of the art in natural product HPLC-MS, HPLC-NMR, and CE-MS hyphenations. Nat. Prod. Rep. 2013, 30, 970–987. [Google Scholar] [CrossRef]
- Gebretsadik, T.; Linert, W.; Thomas, M.; Berhanu, T.; Frew, R. LC-NMR for Natural Products Analysis: A Journey from an Academic Curiosity to a Robust Analytical Tool. Sci 2019, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Keifer, P.A. Chemical-shift referencing and resolution stability in methanol:water gradient LC–NMR. J. Magn. Reson. 2010, 205, 130–140. [Google Scholar] [CrossRef]
- Bringmann, G.; Wohlfarth, M.; Rischer, H.; Heubes, M.; Saeb, W.; Diem, S.; Herderich, M.; Schlauer, J. A Photometric Screening Method for Dimeric Naphthylisoquinoline Alkaloids and Complete On-Line Structural Elucidation of a Dimer in Crude Plant Extracts, by the LC−MS/LC−NMR/LC−CD Triad. Anal. Chem. 2001, 73, 2571–2577. [Google Scholar] [CrossRef]
- Waridel, P.; Wolfender, J.-L.; Lachavanne, J.-B.; Hostettmann, K. ent-Labdane glycosides from the aquatic plant Potamogeton lucens and analytical evaluation of the lipophilic extract constituents of various Potamogeton species. Phytochemistry 2004, 65, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Zehl, M.; Braunberger, C.; Conrad, J.; Crnogorac, M.; Krasteva, S.; Vogler, B.; Beifuss, U.; Krenn, L. Identification and quantification of flavonoids and ellagic acid derivatives in therapeutically important Drosera species by LC–DAD, LC–NMR, NMR, and LC–MS. Anal. Bioanal. Chem. 2011, 400, 2565–2576. [Google Scholar] [CrossRef]
- Clarkson, C.; Sibum, M.; Mensen, R.; Jaroszewski, J.W. Evaluation of on-line solid-phase extraction parameters for hyphenated, high-performance liquid chromatography–solid-phase extraction–nuclear magnetic resonance applications. J. Chromatogr. A 2007, 1165, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, L.; Horton, R. Optimization of LC–NMR. III—Increased signal-to-noise ratio through column trapping. Magn. Reson. Chem. 1998, 36, 104–109. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Capistrano, R.; Dhooghe, L.; Foubert, K.; Lemière, F.; Maregesi, S.; Baldé, A.; Apers, S.; Pieters, L. Herbal Medicines and Infectious Diseases: Characterization by LC-SPE-NMR of Some Medicinal Plant Extracts Used against Malaria. Planta Med. 2011, 77, 1139–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, A.; Moco, S.; Coll, J.; Vervoort, J. LC-MS-SPE-NMR for the Isolation and Characterization of neo-Clerodane Diterpenoids from Teucrium luteum subsp. flavovirens. J. Nat. Prod. 2010, 73, 962–965. [Google Scholar] [CrossRef]
- Gao, H.; Zehl, M.; Kaehlig, H.; Schneider, P.; Stuppner, H.; Moreno, Y.; Banuls, L.; Kiss, R.; Kopp, B. Rapid Structural Identification of Cytotoxic Bufadienolide Sulfates in Toad Venom from Bufo melanosticus by LC-DAD-MSn and LC-SPE-NMR. J. Nat. Prod. 2010, 73, 603–608. [Google Scholar] [CrossRef]
- Chen, C.-K.; Lin, F.-H.; Tseng, L.-H.; Jiang, C.-L.; Lee, S.-S. Comprehensive Study of Alkaloids from Crinum asiaticum var. sinicum Assisted by HPLC-DAD-SPE-NMR. J. Nat. Prod. 2011, 74, 411–419. [Google Scholar] [CrossRef]
- Lai, Y.-C.; Chen, C.-K.; Lin, W.-W.; Lee, S.-S. A comprehensive investigation of anti-inflammatory diarylheptanoids from the leaves of Alnus formosana. Phytochemistry 2012, 73, 84–94. [Google Scholar] [CrossRef]
- Clarkson, C.; Hansen, S.H.; Smith, P.J.; Jaroszewski, J.W. Identification of Major and Minor Constituents of Harpagophytum procumbens (Devil’s Claw) Using HPLC-SPE-NMR and HPLC-ESIMS/APCIMS. J. Nat. Prod. 2006, 69, 1280–1288. [Google Scholar] [CrossRef]
- Nyberg, N.T.; Baumann, H.; Kenne, L. Solid-Phase Extraction NMR Studies of Chromatographic Fractions of Saponins from Quillaja saponaria. Anal. Chem. 2003, 75, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kuhlisch, C.; Pohnert, G. Metabolomics in chemical ecology. Nat. Prod. Rep. 2015, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongstad, K.T.; Özdemir, C.; Barzak, A.; Wubshet, S.G.; Staerk, D. Combined Use of High-Resolution α-Glucosidase Inhibition Profiling and High-Performance Liquid Chromatography–High-Resolution Mass Spectrometry–Solid-Phase Extraction–Nuclear Magnetic Resonance Spectroscopy for Investigation of Antidiabetic Principles in Crude Plant Extracts. J. Agric. Food Chem. 2015, 63, 2257–2263. [Google Scholar] [CrossRef] [PubMed]
- Agnolet, S.; Jaroszewski, J.W.; Verpoorte, R.; Staerk, D. 1H NMR-based metabolomics combined with HPLC-PDA-MS-SPE-NMR for investigation of standardized Ginkgo biloba preparations. Metabolomics 2010, 6, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Xiao, C.; Wang, Y. Important roles of the hyphenated HPLC-DAD-MS-SPE-NMR technique in metabonomics. Magn. Reson. Chem. 2009, 47, S157–S162. [Google Scholar] [CrossRef] [PubMed]
- Staerk, D.; Kesting, J.R.; Sairafianpour, M.; Witt, M.; Asili, J.; Emami, S.A.; Jaroszewski, J.W. Accelerated dereplication of crude extracts using HPLC–PDA–MS–SPE–NMR: Quinolinone alkaloids of Haplophyllum acutifolium. Phytochemistry 2009, 70, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, R.d.C.L.; Gramsbergen, S.M.; Van Staden, J.; Jäger, A.K.; Kongstad, K.T.; Staerk, D. Advancing HPLC-PDA-HRMS-SPE-NMR Analysis of Coumarins in Coleonema album by Use of Orthogonal Reversed-Phase C18 and Pentafluorophenyl Separations. J. Nat. Prod. 2017, 80, 1020–1027. [Google Scholar] [CrossRef]
- Liu, B.; Kongstad, K.T.; Qinglei, S.; Nyberg, N.T.; Jäger, A.K.; Staerk, D. Dual High-Resolution α-Glucosidase and Radical Scavenging Profiling Combined with HPLC-HRMS-SPE-NMR for Identification of Minor and Major Constituents Directly from the Crude Extract of Pueraria lobata. J. Nat. Prod. 2015, 78, 294–300. [Google Scholar] [CrossRef]
- Luedemann, A.; Strassburg, K.; Erban, A.; Kopka, J. TagFinder for the quantitative analysis of gas chromatography—Mass spectrometry (GC-MS)-based metabolite profiling experiments. Bioinformatics 2008, 24, 732–737. [Google Scholar] [CrossRef]
- Cuadros-Inostroza, Á.; Caldana, C.; Redestig, H.; Kusano, M.; Lisec, J.; Peña-Cortés, H.; Willmitzer, L.; Hannah, M.A. TargetSearch—A Bioconductor package for the efficient preprocessing of GC-MS metabolite profiling data. BMC Bioinform. 2009, 10, 428. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Tsugawa, H.; Miyagawa, H.; Fukusaki, E. Integrated Strategy for Unknown EI–MS Identification Using Quality Control Calibration Curve, Multivariate Analysis, EI–MS Spectral Database, and Retention Index Prediction. Anal. Chem. 2017, 89, 6766–6773. [Google Scholar] [CrossRef] [PubMed]
- Beale, D.J.; Pinu, F.R.; Kouremenos, K.A.; Poojary, M.M.; Narayana, V.K.; Boughton, B.A.; Kanojia, K.; Dayalan, S.; Jones, O.A.H.; Dias, D.A. Review of recent developments in GC–MS approaches to metabolomics-based research. Metabolomics 2018, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics by Gas Chromatography—Mass Spectrometry: Combined Targeted and Untargeted Profiling. Curr. Protoc. Mol. Biol. 2016, 114, 30.4.1–30.4.32. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Schiffman, C.; Petrick, L.; Perttula, K.; Yano, Y.; Carlsson, H.; Whitehead, T.; Metayer, C.; Hayes, J.; Rappaport, S.; Dudoit, S. Filtering procedures for untargeted LC-MS metabolomics data. BMC Bioinform. 2019, 20, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.-F.; Nothias-Esposito, M.; da Silva, R.; Wang, M.; Protsyuk, I.; Zhang, Z.; Sarvepalli, A.; Leyssen, P.; Touboul, D.; Costa, J.; et al. Bioactivity-Based Molecular Networking for the Discovery of Drug Leads in Natural Product Bioassay-Guided Fractionation. J. Nat. Prod. 2018, 81, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.-J. MetDIA: Targeted Metabolite Extraction of Multiplexed MS/MS Spectra Generated by Data-Independent Acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.-L.; Nuzillard, J.-M.; van der Hooft, J.J.J.; Renault, J.-H.; Bertrand, S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography–High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019, 91, 704–742. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2017, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Manach, C.; Andres-Lacueva, C.; Dragsted, L.O.; Draper, J.; Rappaport, S.M.; van der Hooft, J.J.; Wishart, D.S. The food metabolome: A window over dietary exposure. Am. J. Clin. Nutr. 2014, 99, 1286–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaženović, I.; Kind, T.; Torbašinović, H.; Obrenović, S.; Mehta, S.S.; Tsugawa, H.; Wermuth, T.; Schauer, N.; Jahn, M.; Biedendieck, R.; et al. Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: Database boosting is needed to achieve 93% accuracy. J. Cheminformatics 2017, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Kind, T.; Nakabayashi, R.; Yukihira, D.; Tanaka, W.; Cajka, T.; Saito, K.; Fiehn, O.; Arita, M. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem. 2016, 88, 7946–7958. [Google Scholar] [CrossRef]
- Stanstrup, J.; Neumann, S.; Vrhovšek, U. PredRet: Prediction of Retention Time by Direct Mapping between Multiple Chromatographic Systems. Anal. Chem. 2015, 87, 9421–9428. [Google Scholar] [CrossRef] [Green Version]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK Family Databases: Integrated Metabolite–Plant Species Databases for Multifaceted Plant Research. Plant Cell Physiol. 2011, 53, e1. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.K.; Boyd, R.K.; Eberlin, M.N.; Langley, G.J.; Li, L.; Naito, Y. Definitions of terms relating to mass spectrometry (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1515–1609. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakabayashi, R.; Yamada, Y.; Suzuki, M.; Sato, M.; Sakata, A.; Akiyama, K.; Sakurai, T.; Matsuda, F.; Aoki, T. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: A plant-specific MS/MS-based data resource and database. Phytochemistry 2012, 82, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohimani, H.; Gurevich, A.; Mikheenko, A.; Garg, N.; Nothias, L.F.; Ninomiya, A.; Takada, K.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 2017, 13, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar] [CrossRef]
- Gowda, G.N.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef] [Green Version]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Lewis, I.A.; Schommer, S.C.; Markley, J.L. rNMR: Open source software for identifying and quantifying metabolites in NMR spectra. Magn. Reson. Chem. 2009, 47, S123–S126. [Google Scholar] [CrossRef]
- Chikayama, E.; Sekiyama, Y.; Okamoto, M.; Nakanishi, Y.; Tsuboi, Y.; Akiyama, K.; Saito, K.; Shinozaki, K.; Kikuchi, J. Statistical indices for simultaneous large-scale metabolite detections for a single NMR spectrum. Anal. Chem. 2010, 82, 1653–1658. [Google Scholar] [CrossRef]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.-W.; Bruschweiler, R. Customized metabolomics database for the analysis of NMR 1H–1H TOCSY and 13C–1H HSQC-TOCSY spectra of complex mixtures. Anal. Chem. 2014, 86, 5494–5501. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Zhang, F.; Bruschweiler-Li, L.; Bruschweiler, R. TOCCATA: A customized carbon total correlation spectroscopy NMR metabolomics database. Anal. Chem. 2012, 84, 9395–9401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Pérez, J.L.; Therón, R.; del Olmo, E.; Díaz, D. NAPROC-13: A database for the dereplication of natural product mixtures in bioassay-guided protocols. Bioinformatics 2007, 23, 3256–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbeck, C.; Krause, S.; Kuhn, S. NMRShiftDB constructing a free chemical information system with open-source components. J. Chem. Inf. Comput. Sci. 2003, 43, 1733–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbeck, C.; Kuhn, S. NMRShiftDB–compound identification and structure elucidation support through a free community-built web database. Phytochemistry 2004, 65, 2711–2717. [Google Scholar] [CrossRef]
- Yamamoto, O.; Someno, K.; Wasada, N.; Hiraishi, J.; Hayamizu, K.; Tanabe, K.; Tamura, T.; Yanagisawa, M. An integrated spectral data base system including IR, MS, 1H-NMR, 13C-NMR, ESR and Raman spectra. Anal. Sci. 1988, 4, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, C.; Easton, J.M.; Lodi, A.; Tiziani, S.; Manzoor, S.E.; Southam, A.D.; Byrne, J.J.; Bishop, L.M.; He, S.; Arvanitis, T.N. Birmingham metabolite library: A publicly accessible database of 1-D 1H and 2-D 1H J-resolved NMR spectra of authentic metabolite standards (BML-NMR). Metabolomics 2012, 8, 8–18. [Google Scholar] [CrossRef]
- Ulrich, E.; Akutsu, H.; Doreleijers, J.; Harano, Y.; Ioannidis, Y.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanshole, V.V.; Snytnikova, O.A.; Kiryutin, A.S.; Yanshole, L.V.; Sagdeev, R.Z.; Tsentalovich, Y.P. Metabolomics of the rat lens: A combined LC-MS and NMR study. Exp. Eye Res. 2014, 125, 71–78. [Google Scholar] [CrossRef]
- Bingol, K.; Bruschweiler-Li, L.; Yu, C.; Somogyi, A.; Zhang, F.; Brüschweiler, R. Metabolomics beyond spectroscopic databases: A combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal. Chem. 2015, 87, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Bingol, K.; Brüschweiler, R. Two elephants in the room: New hybrid nuclear magnetic resonance and mass spectrometry approaches for metabolomics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 471. [Google Scholar] [CrossRef]
- Wohlleben, W.; Mast, Y.; Muth, G.; Röttgen, M.; Stegmann, E.; Weber, T. Synthetic biology of secondary metabolite biosynthesis in actinomycetes: Engineering precursor supply as a way to optimize antibiotic production. FEBS Lett. 2012, 586, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Jankevics, A.; Merlo, M.E.; de Vries, M.; Vonk, R.J.; Takano, E.; Breitling, R. Metabolomic analysis of a synthetic metabolic switch in Streptomyces coelicolor A3 (2). Proteomics 2011, 11, 4622–4631. [Google Scholar] [CrossRef] [PubMed]
- Breitling, R.; Achcar, F.; Takano, E. Modeling challenges in the synthetic biology of secondary metabolism. ACS Synth. Biol. 2013, 2, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-S.; Liang, Y.-Q.; Ding, M.-Z.; Cui, S.-F.; Lv, X.-M.; Yuan, Y.-J. Metabolic analysis reveals the amino acid responses of Streptomyces lydicus to pitching ratios during improving streptolydigin production. Appl. Microbiol. Biotechnol. 2013, 97, 5943–5954. [Google Scholar] [CrossRef]
- Arendt, P.; Pollier, J.; Callewaert, N.; Goossens, A. Synthetic biology for production of natural and new-to-nature terpenoids in photosynthetic organisms. Plant J. Cell Mol. Biol. 2016, 87, 16–37. [Google Scholar] [CrossRef] [Green Version]
- Julsing, M.K.; Koulman, A.; Woerdenbag, H.J.; Quax, W.J.; Kayser, O. Combinatorial biosynthesis of medicinal plant secondary metabolites. Biomol. Eng. 2006, 23, 265–279. [Google Scholar] [CrossRef]
- Pollier, J.; Moses, T.; Goossens, A. Combinatorial biosynthesis in plants: A (p)review on its potential and future exploitation. Nat. Prod. Rep. 2011, 28, 1897–1916. [Google Scholar] [CrossRef]
- Umeno, D.; Arnold, F.H. A C35 carotenoid biosynthetic pathway. Appl. Environ. Microbiol. 2003, 69, 3573–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runguphan, W.; Qu, X.; O’Connor, S.E. Integrating carbon-halogen bond formation into medicinal plant metabolism. Nature 2010, 468, 461–464. [Google Scholar] [CrossRef]
- Moses, T.; Papadopoulou, K.K.; Osbourn, A. Metabolic and functional diversity of saponins, biosynthetic intermediates and semi-synthetic derivatives. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 439–462. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, P.; Zhou, F.; Erban, A.; Karcher, D.; Kopka, J.; Bock, R. A new synthetic biology approach allows transfer of an entire metabolic pathway from a medicinal plant to a biomass crop. eLife 2016, 5, e13664. [Google Scholar] [CrossRef]
- Hiller, K.; Metallo, C.M.; Kelleher, J.K.; Stephanopoulos, G. Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Anal. Chem. 2010, 82, 6621–6628. [Google Scholar] [CrossRef] [PubMed]
- Creek, D.J.; Chokkathukalam, A.; Jankevics, A.; Burgess, K.E.; Breitling, R.; Barrett, M.P. Stable isotope-assisted metabolomics for network-wide metabolic pathway elucidation. Anal. Chem. 2012, 84, 8442–8447. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Krska, R.; Kluger, B.; Schuhmacher, R. Isotopic labeling-assisted metabolomics using LC–MS. Anal. Bioanal. Chem. 2013, 405, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, D.I.; Goodacre, R. Metabolomics-assisted synthetic biology. Curr. Opin. Biotechnol. 2012, 23, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, J.; Chen, X.H.; Borriss, R.; Piel, J. Biosynthesis of the antibiotic bacillaene, the product of a giant polyketide synthase complex of the trans-AT family. Angew. Chem. Int. Ed. 2007, 46, 8195–8197. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Munro, M.H. Data, 1 H-NMR databases, data manipulation. Phytochem. Rev. 2013, 12, 435–447. [Google Scholar] [CrossRef]
- Buckingham, J. Dictionary of Natural Products on DVD; Version 21: 1; Chapman & Hall/CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Purves, K.; Macintyre, L.; Brennan, D.; Hreggviðsson, G.; Kuttner, E.; Ásgeirsdóttir, M.; Young, L.; Green, D.; Edrada-Ebel, R.; Duncan, K. Using molecular networking for microbial secondary metabolite bioprospecting. Metabolites 2016, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Funasaki, M.; Menezes, I.S.; dos Santos BARROSO, H.; Zanotto, S.P.; Carioca, C.R.F. Tocopherol profile of Brazil nut oil from different geographic areas of the Amazon region. Acta Amaz. 2012, 43. [Google Scholar] [CrossRef] [Green Version]
- Borges, C.V.; de Oliveira Amorim, V.B.; Ramlov, F.; da Silva Ledo, C.A.; Donato, M.; Maraschin, M.; Amorim, E.P. Characterisation of metabolic profile of banana genotypes, aiming at biofortified Musa spp. cultivars. Food Chem. 2014, 145, 496–504. [Google Scholar] [CrossRef] [PubMed]
- da Silva Taveira, J.H.; Borém, F.M.; Figueiredo, L.P.; Reis, N.; Franca, A.S.; Harding, S.A.; Tsai, C.-J. Potential markers of coffee genotypes grown in different Brazilian regions: A metabolomics approach. Food Res. Int. 2014, 61, 75–82. [Google Scholar] [CrossRef]
- Pilon, A.C.; Carnevale Neto, F.; Freire, R.T.; Cardoso, P.; Carneiro, R.L.; Da Silva Bolzani, V.; Castro-Gamboa, I. Partial least squares model and design of experiments toward the analysis of the metabolome of Jatropha gossypifolia leaves: Extraction and chromatographic fingerprint optimization. J. Sep. Sci. 2016, 39, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.M.; Bergamasco, J.; Lira, S.P.; Lopes, N.P.; Hajdu, E.; Peixinho, S.; Berlinck, R.G. Dereplication of bromotyrosine-derived metabolites by LC-PDA-MS and analysis of the chemical profile of 14 aplysina sponge specimens from the Brazilian coastline. Aust. J. Chem. 2010, 63, 886–894. [Google Scholar] [CrossRef]
- Bittencourt, M.L.; Ribeiro, P.R.; Franco, R.L.; Hilhorst, H.W.; de Castro, R.D.; Fernandez, L.G. Metabolite profiling, antioxidant and antibacterial activities of Brazilian propolis: Use of correlation and multivariate analyses to identify potential bioactive compounds. Food Res. Int. 2015, 76, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, C.B.; Pires, D.O. Brazilian coral reefs: What we already know and what is still missing. Bull. Mar. Sci. 2001, 69, 357–371. [Google Scholar]
- Funari, C.S.; Eugster, P.J.; Martel, S.; Carrupt, P.-A.; Wolfender, J.-L.; Silva, D.H.S. High resolution ultra high pressure liquid chromatography–time-of-flight mass spectrometry dereplication strategy for the metabolite profiling of Brazilian Lippia species. J. Chromatogr. A 2012, 1259, 167–178. [Google Scholar] [CrossRef]
- Chagas-Paula, D.A.; Oliveira, T.B.; Zhang, T.; Edrada-Ebel, R.; Da Costa, F.B. Prediction of anti-inflammatory plants and discovery of their biomarkers by machine learning algorithms and metabolomic studies. Planta Med. 2015, 81, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Nicholson, J.K.; Hylands, P.J.; Sampson, J.; Whitcombe, I.; Stewart, C.G.; Caiger, S.; Oru, I.; Holmes, E. Metabolomic strategy for the classification and quality control of phytomedicine: A case study of chamomile flower (Matricaria recutita L.). Planta Med. 2004, 70, 250–255. [Google Scholar]
- Choi, Y.H.; Kim, H.K.; Hazekamp, A.; Erkelens, C.; Lefeber, A.W.; Verpoorte, R. Metabolomic Differentiation of Cannabis s ativa Cultivars Using 1H NMR Spectroscopy and Principal Component Analysis. J. Nat. Prod. 2004, 67, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.; Cloarec, O.; Tang, H.; Stærk, D.; Jaroszewski, J.W. Multivariate analysis of integrated and full-resolution 1H-NMR spectral data from complex pharmaceutical preparations: St. John’s wort. Planta Med. 2006, 72, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kooy, F.; Verpoorte, R.; Meyer, J.M. Metabolomic quality control of claimed anti-malarial Artemisia afra herbal remedy and A. afra and A. annua plant extracts. S. Afr. J. Bot. 2008, 74, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Zhang, J.-L.; Li, F. Application of Metabolomics in the Study of Natural Products. Nat. Prod. Bioprospecting 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
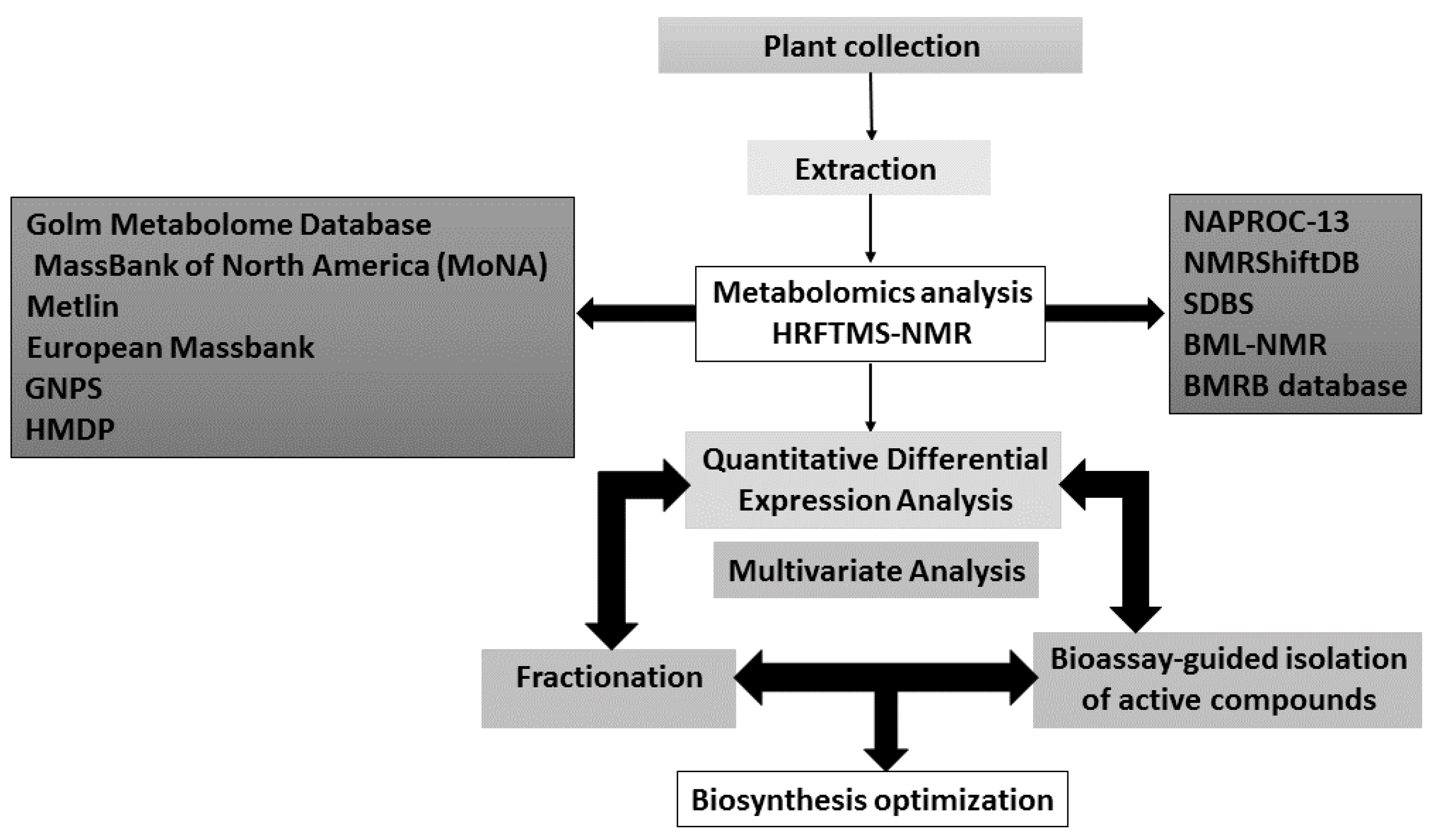
| Database | Applications | Notes | References |
|---|---|---|---|
| NAPROC-13 | Is useful in cases of dereplication as it has more than 20.000 of natural product spectra and inclusion of metabolite classification | Its closed design, there is no user access to spectral data. 13C-NMR spectra, molecular formula or predicted molecular weight can only be searched separately. | [211] |
| NMRShiftDB | Not limited to NP and has 51,000 collected spectra. It accepts submissions | It lists NMR chemical shifts, but not peak size. | [212,213] |
| SDBS | Not limited to NP and has 29,000 collected spectra. It uses multiple kinds of spectra in a single search. | Does not accept submissions. | [214] |
| BML-NMR | The spectral depth of 16 different one- and two-dimensional experiments for each compound provides high quality references | Covers only 208 compounds, but each compound is measured with 16 different NMR parameter sets which provides high quality references. These metabolites were selected based on their importance within metabolic pathways and their detection potential by NMR. They were analyzed at pH 6.6, 7.0, and 7.4. | [215] |
| BMRB database | Contains NMR data for various biomolecules with a focus on protein, peptide, and nucleic acid spectra. Spectra are available for downloading in raw and processed data formats | The database mainly covers the compounds involved in the lignin biosynthesis which is a plant cell wall constituent and the compounds obtained by plant cell wall deconstruction. | [216] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, M.A.; Perez de Souza, L.; Serag, A.; Fernie, A.R.; Farag, M.A.; Ezzat, S.M.; Alseekh, S. Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis. Metabolites 2020, 10, 37. https://doi.org/10.3390/metabo10010037
Salem MA, Perez de Souza L, Serag A, Fernie AR, Farag MA, Ezzat SM, Alseekh S. Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis. Metabolites. 2020; 10(1):37. https://doi.org/10.3390/metabo10010037
Chicago/Turabian StyleSalem, Mohamed A., Leonardo Perez de Souza, Ahmed Serag, Alisdair R. Fernie, Mohamed A. Farag, Shahira M. Ezzat, and Saleh Alseekh. 2020. "Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis" Metabolites 10, no. 1: 37. https://doi.org/10.3390/metabo10010037
APA StyleSalem, M. A., Perez de Souza, L., Serag, A., Fernie, A. R., Farag, M. A., Ezzat, S. M., & Alseekh, S. (2020). Metabolomics in the Context of Plant Natural Products Research: From Sample Preparation to Metabolite Analysis. Metabolites, 10(1), 37. https://doi.org/10.3390/metabo10010037