Developmental Programming and Glucolipotoxicity: Insights on Beta Cell Inflammation and Diabetes


Abstract
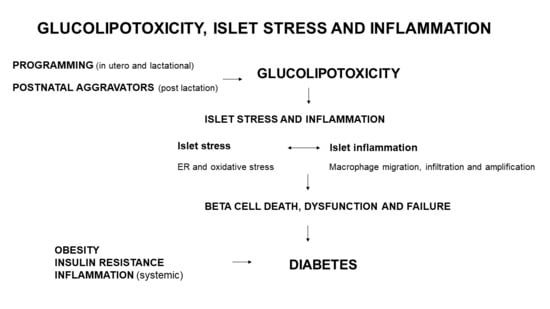
:
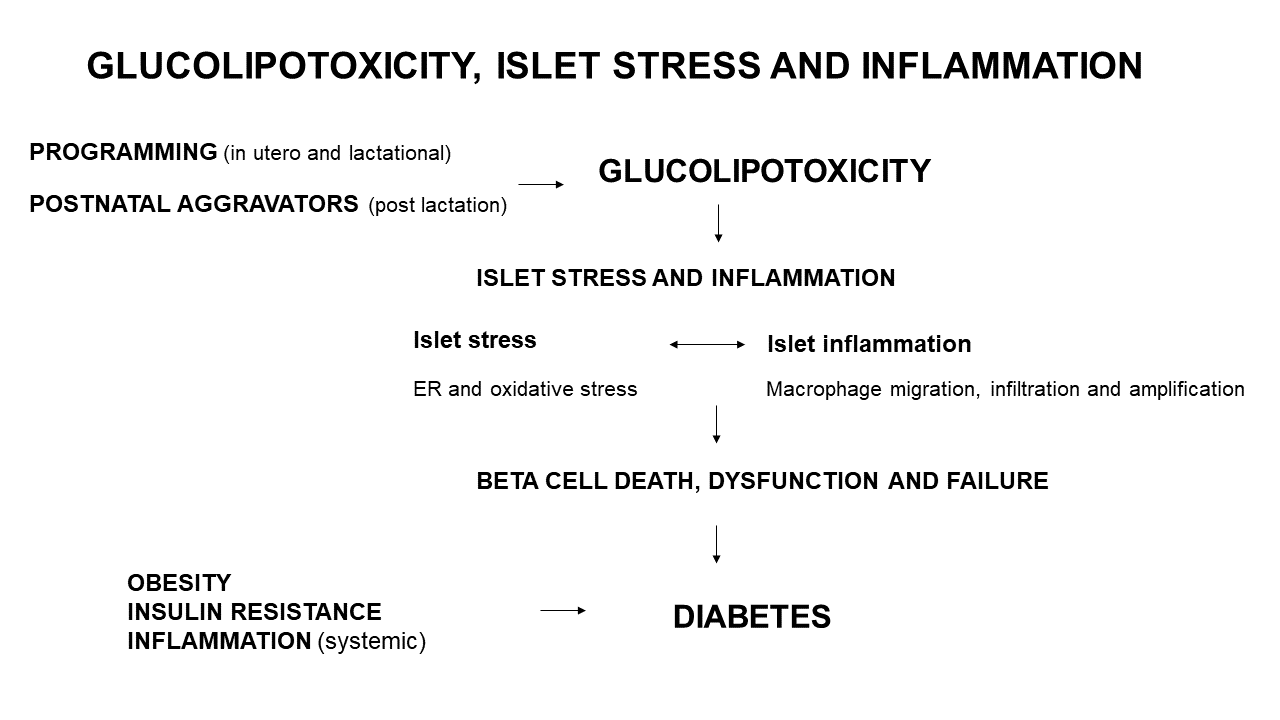{kind=link}
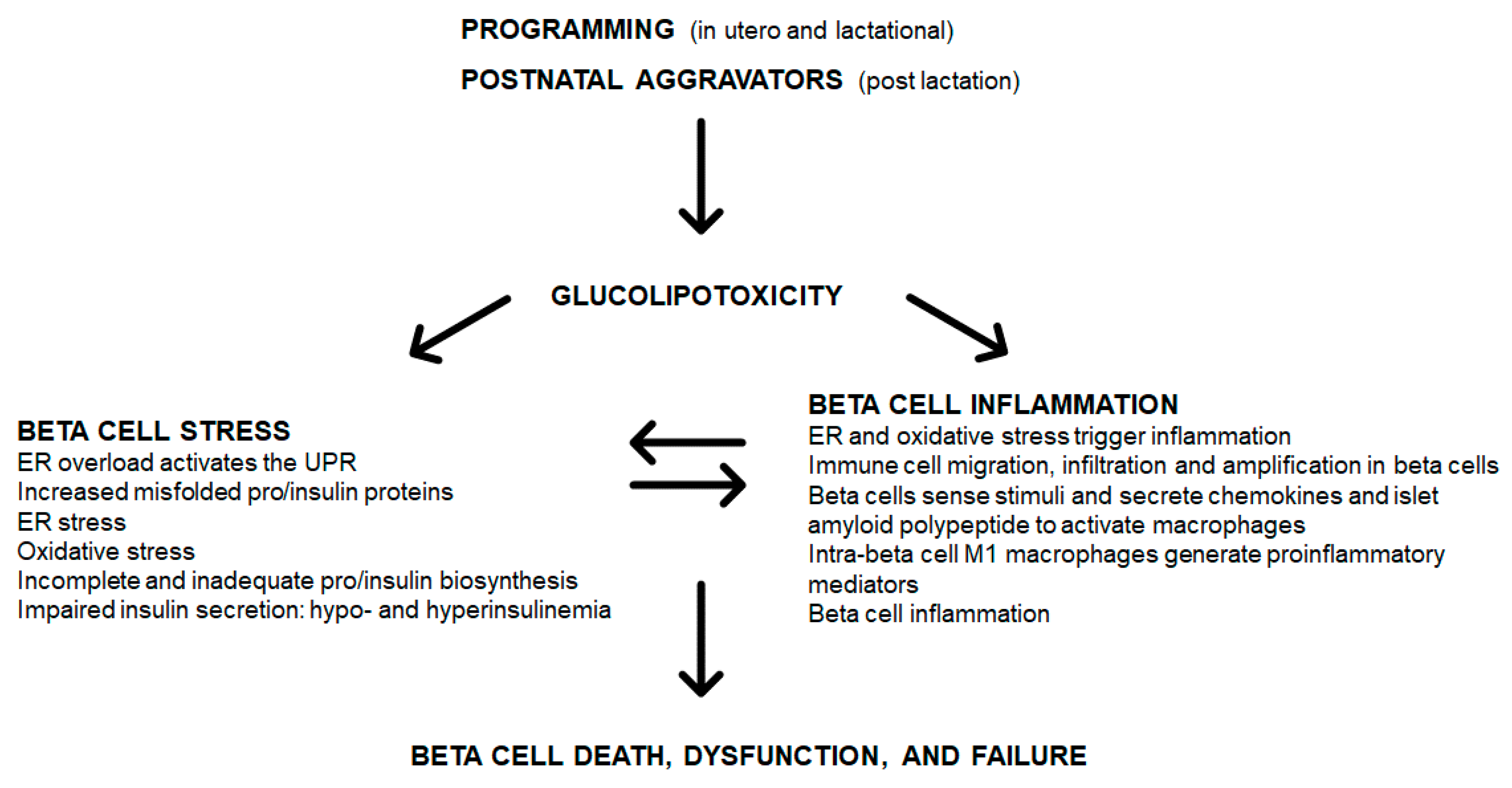{kind=link}
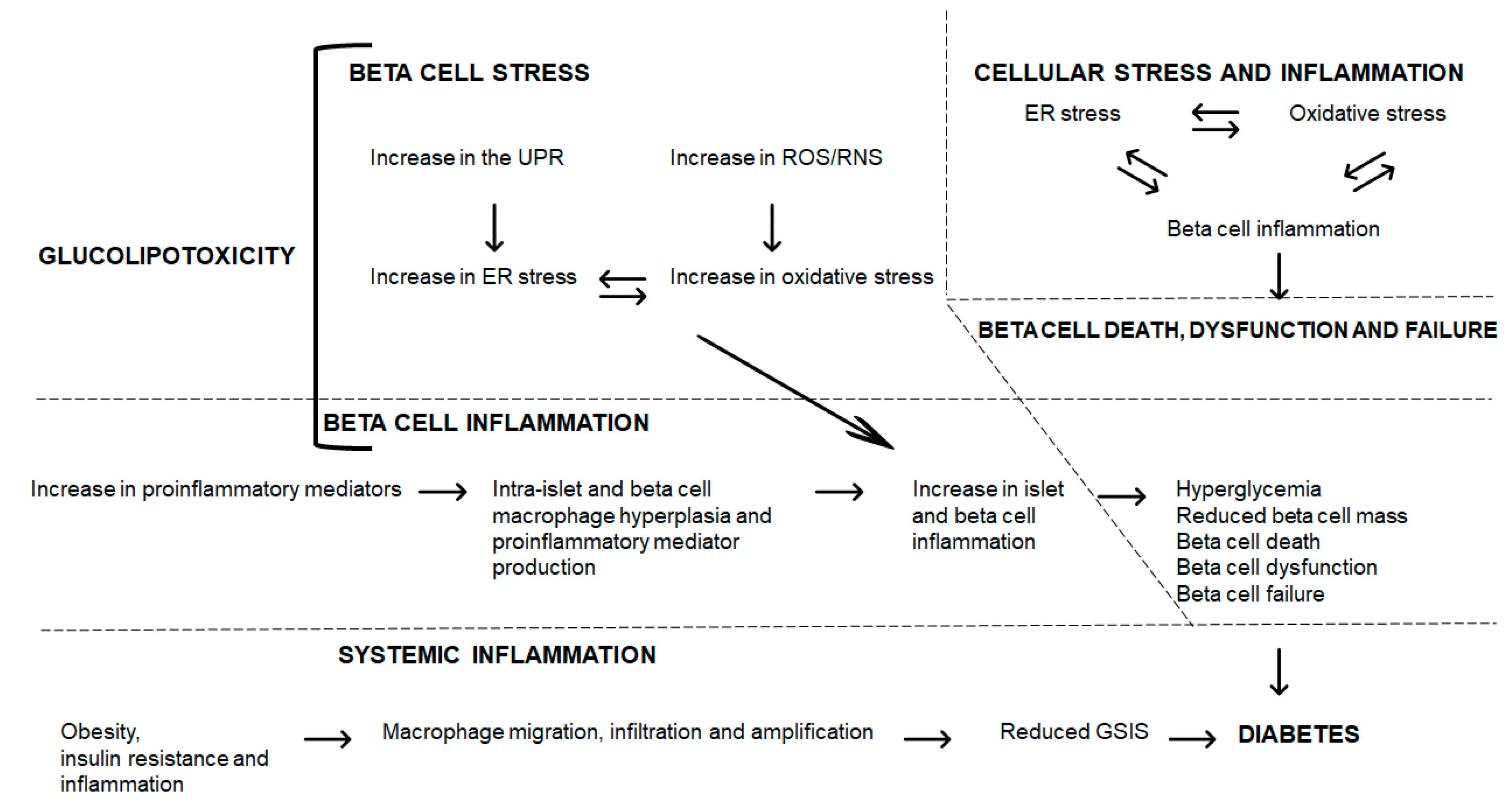{kind=link}
1. Introduction
2. Islet Inflammation
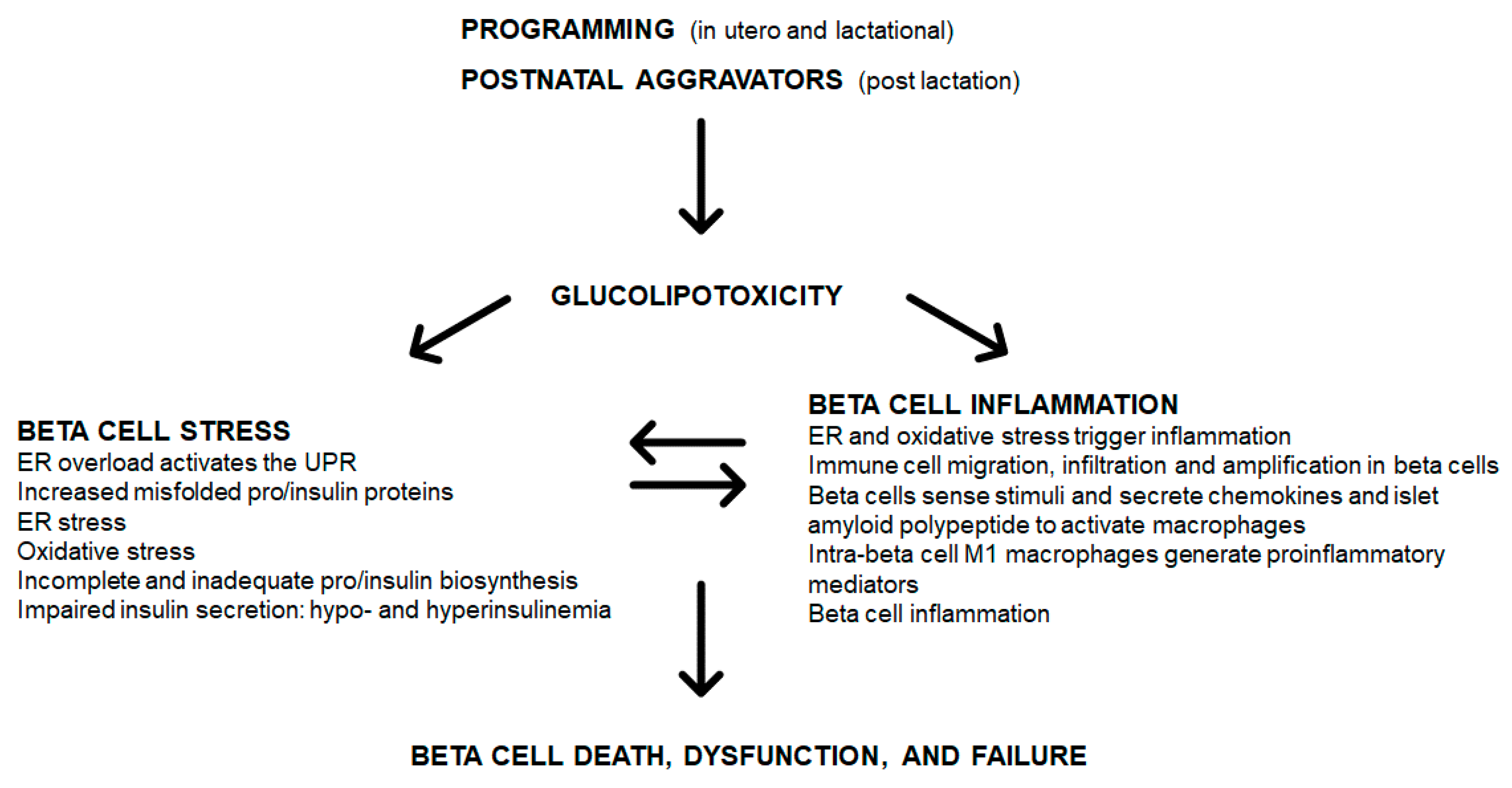
3. Programming and Islet Inflammation
4. Glucolipotoxicity
4.1. Overview
4.2. Islet and Beta Cell Glucolipotoxicity, Stress and Inflammation
5. ER and Oxidative Stress, Inflammation and Beta Cell Dysfunction
5.1. ER Stress
5.2. Oxidative Stress
5.3. Beta Cell Stress and Inflammation
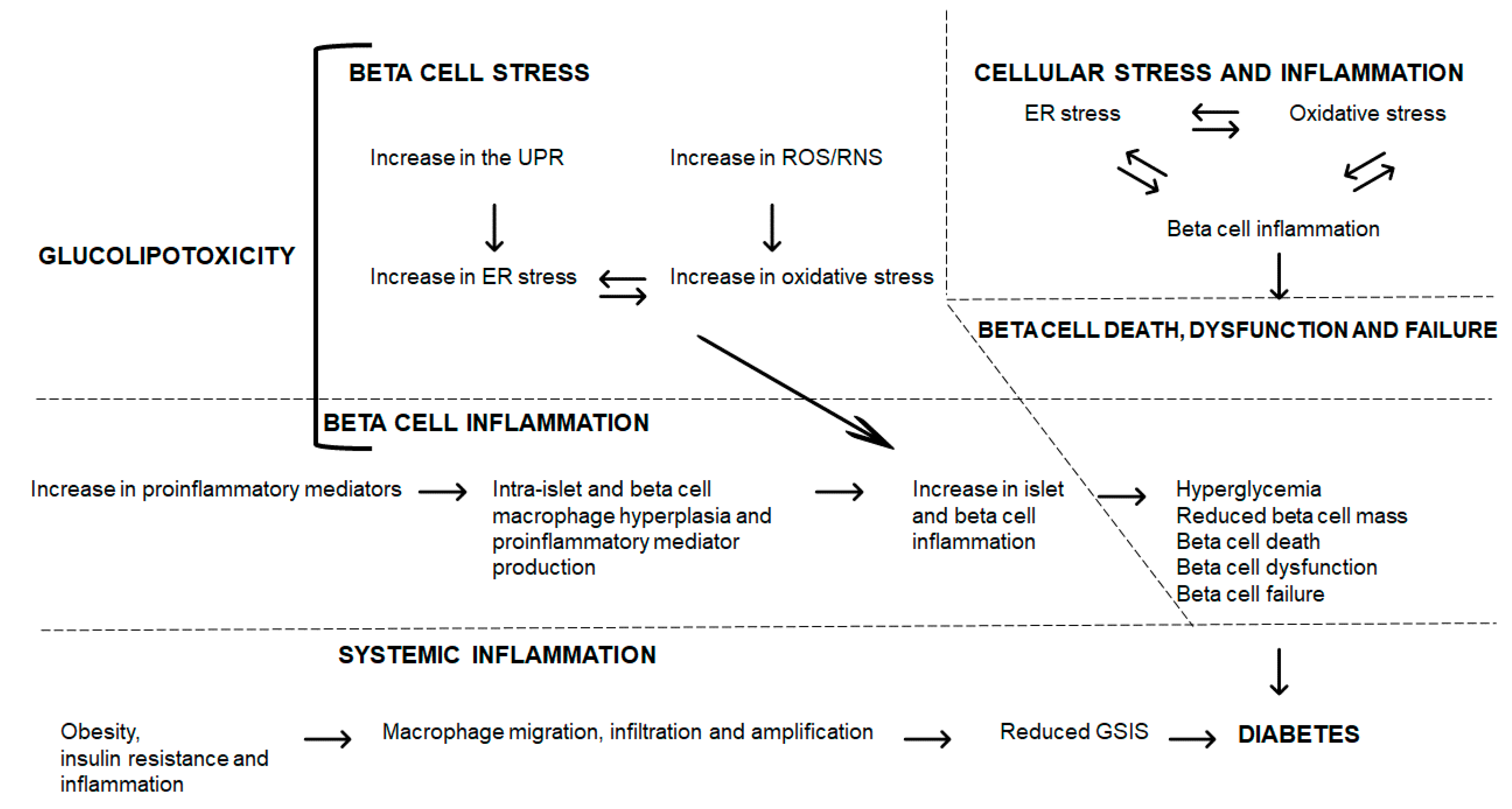
6. Glucolipotoxicity: Interrelating Islet and Beta Cell Stress, Inflammation to Dysfunction
7. Conclusions
Funding
Conflicts of Interest
References
- Cerf, M.E.; Williams, K.; Nkomo, X.I.; Muller, C.J.; Du Toit, D.F.; Louw, J.; Wolfe-Coote, S.A. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1122–R1128. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E.; Chapman, C.S.; Muller, C.J.; Louw, J. Gestational high-fat programming impairs insulin release and reduces Pdx-1 and glucokinase immunoreactivity in neonatal Wistar rats. Metabolism 2009, 58, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E.; Muller, C.J.; Du Toit, D.F.; Louw, J.; Wolfe-Coote, S.A. Hyperglycaemia and reduced glucokinase expression in weanling offspring from dams maintained on a high-fat diet. Br. J. Nutr. 2006, 95, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerf, M.E.; Louw, J. Islet cell response to high fat programming in neonate, weanling and adolescent Wistar rats. JOP 2014, 15, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Gniuli, D.; Calcagno, A.; Caristo, M.E.; Mancuso, A.; Macchi, V.; Mingrone, G.; Vettor, R. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J. Lipid Res. 2008, 49, 1936–1945. [Google Scholar] [CrossRef] [Green Version]
- Karbaschi, R.; Zardooz, H.; Khodagholi, F.; Dargahi, L.; Salimi, M.; Rashidi, F. Maternal high-fat diet intensifies the metabolic response to stress in male rat offspring. Nutr. Metab. 2017, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. A maternal high-fat diet induces DNA methylation changes that contribute to glucose intolerance in offspring. Front. Endocrinol. 2019, 10, 871. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, E.; Sosa-Larios, T.; Calzada, L.; Ibáñez, C.A.; Mendoza-Rodríguez, C.A.; Morales, A.; Morimoto, S. Decreased basal insulin secretion from pancreatic islets of pups in a rat model of maternal obesity. J. Endocrinol. 2016, 231, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Cerf, M.E.; Louw, J.; Herrera, E. High fat diet exposure during fetal life enhances plasma and hepatic omega-6 fatty acid profiles in fetal Wistar rats. Nutrients 2015, 7, 7231–7241. [Google Scholar] [CrossRef] [Green Version]
- Cerf, M.E.; Herrera, E. High fat diet administration during specific periods of pregnancy alters maternal fatty acid profiles in the near-term rat. Nutrients 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; De Bittencourt, P.I.H.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and β-cell dysfunction. Oxid. Med. Cell. Longev. 2015, 29, 457–474.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [Green Version]
- Böni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, H.; Inoguchi, T.; Sonoda, N.; Sakaki, Y.; Maeda, Y.; Inoue, T.; Hirata, E.; Takei, R.; Ikeda, N.; Fujii, M.; et al. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic β-cell function in adult offspring with sex differences in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1163–E1175. [Google Scholar] [CrossRef] [Green Version]
- Nicol, L.E.; Grant, W.F.; Comstock, S.M.; Nguyen, M.L.; Smith, M.S.; Grove, K.L.; Marks, D.L. Pancreatic inflammation and increased islet macrophages in insulin-resistant juvenile primates. J. Endocrinol. 2013, 217, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Marselli, L.; Thorne, J.; Dahiya, S.; Sgroi, D.C.; Sharma, A.; Bonner-Weir, S.; Marchetti, P.; Weir, G.C. Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PLoS ONE 2010, 5, e11499. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Cao, H.; Du, J.; Liu, X.; Liu, Y.; Xiang, M. Reg3g overexpression promotes β cell regeneration and induces immune tolerance in non-obese diabetic mouse model. J. Leukoc. Biol. 2016, 99, 1131–1140. [Google Scholar] [CrossRef] [Green Version]
- Casasnovas, J.; Jo, Y.; Rao, X.; Xuei, X.; Brown, M.E.; Kua, K.L. High glucose alters fetal rat islet transcriptome and induces progeny islet dysfunction. J. Endocrinol. 2019, 240, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Khaodhiar, L.; Ling, P.R.; Blackburn, G.L.; Bistrian, B.R. Serum levels of interleukin-6 and C-reactive protein correlate with body mass index across the broad range of obesity. J. Parenter. Enter. Nutr. 2004, 28, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Is obesity an inflammatory condition? Nutrition 2001, 17, 953–966. [Google Scholar] [CrossRef]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Catalano, P.M.; Presley, L.; Minium, J.; Hauguel-de Mouzon, S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care 2009, 32, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Madan, J.C.; Davis, J.M.; Craig, W.Y.; Collins, M.; Allan, W.; Quinn, R.; Dammann, O. Maternal obesity and markers of inflammation in pregnancy. Cytokine 2009, 47, 61–64. [Google Scholar] [CrossRef]
- Roberts, K.A.; Riley, S.C.; Reynolds, R.M.; Barr, S.; Evans, M.; Statham, A.; Hor, K.; Jabbour, H.N.; Norman, J.E.; Denison, F.C. Placental structure and inflammation in pregnancies associated with obesity. Placenta 2011, 32, 247–254. [Google Scholar] [CrossRef]
- Keȩpczyńska, M.A.; Wargent, E.T.; Cawthorne, M.A.; Arch, J.R.S.; O’Dowd, J.F.; Stocker, C.J. Circulating levels of the cytokines IL10, IFNγ and resistin in an obese mouse model of developmental programming. J. Dev. Orig. Health Dis. 2013, 4, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef]
- Dahlgren, J.; Samuelsson, A.M.; Jansson, T.; Holmäng, A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr. Res. 2006, 60, 147–151. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.J.; Du, M.; Nathanielsz, P.W.; Ford, S.P. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 2010, 31, 387–391. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Hauguel-de Mouzon, S.; Guerre-Millo, M. The placenta cytokine network and inflammatory signals. Placenta 2006, 27, 794–798. [Google Scholar] [CrossRef]
- Aaltonen, R.; Heikkinen, T.; Hakala, K.; Laine, K.; Alanen, A. Transfer of proinflammatory cytokines across term placenta. Obstet. Gynecol. 2005, 106, 802–807. [Google Scholar] [CrossRef]
- Kim, D.W.; Young, S.L.; Grattan, D.R.; Jasoni, C.L. Obesity during pregnancy disrupts placental morphology, cell proliferation, and inflammation in a sex-specific manner across gestation in the mouse. Biol. Reprod. 2014, 90, 130. [Google Scholar] [CrossRef] [Green Version]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Assaad, W.; Buteau, J.; Peyot, M.L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic β-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Somesh, B.P.; Verma, M.K.; Sadasivuni, M.K.; Mammen-Oommen, A.; Biswas, S.; Shilpa, P.C.; Reddy, A.K.; Yateesh, A.N.; Pallavi, P.M.; Nethra, S.; et al. Chronic glucolipotoxic conditions in pancreatic islets impair insulin secretion due to dysregulated calcium dynamics, glucose responsiveness and mitochondrial activity. BMC Cell Biol. 2013, 14, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Barlow, J.; Affourtit, C. Novel insights into pancreatic β-cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells. Biochem. J. 2013, 456, 417–426. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, β-cells, and diabetes: The emperor has no clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Murthy Madiraju, S.R. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef]
- Pinnick, K.; Neville, M.; Clark, A.; Fielding, B. Reversibility of metabolic and morphological changes associated with chronic exposure of pancreatic islet β-cells to fatty acids. J. Cell. Biochem. 2010, 109, 683–692. [Google Scholar] [CrossRef]
- Hu, H.Q.; Qiao, J.T.; Liu, F.Q.; Wang, J.B.; Sha, S.; He, Q.; Cui, C.; Song, J.; Zang, N.; Wang, L.S.; et al. The STING-IRF3 pathway is involved in lipotoxic injury of pancreatic β cells in type 2 diabetes. Mol. Cell. Endocrinol. 2020, 11089. [Google Scholar] [CrossRef]
- Lytrivi, M.; Ghaddar, K.; Lopes, M.; Rosengren, V.; Piron, A.; Yi, X.; Johansson, H.; Lehtiö, J.; Igoillo-Esteve, M.; Cunha, D.A.; et al. Combined transcriptome and proteome profiling of the pancreatic β-cell response to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genom. 2020, 21, 590. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cnop, M.; Ladrière, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic β-cell dysfunction. Diabetes Obes. Metab. 2010, 12, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnain, S.Z.; Borg, D.J.; Harcourt, B.E.; Tong, H.; Sheng, Y.H.; Ng, C.P.; Das, I.; Wang, R.; Chen, A.C.; Loudovaris, T.; et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat. Med. 2014, 20, 1417–1426. [Google Scholar] [CrossRef]
- Elouil, H.; Bensellam, M.; Guiot, Y.; Vander Mierde, D.; Pascal, S.M.; Schuit, F.C.; Jonas, J.C. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 2007, 50, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [Green Version]
- Böni-Schnetzler, M.; Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-Conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; et al. Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef]
- Van Raalte, D.H.; Diamant, M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: Target for durable therapy? Diabetes Res. Clin. Pract. 2011, 93, S37–S46. [Google Scholar] [CrossRef]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Nadler, J.L. Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol. Metab. 2013, 24, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Gross, D.J.; Cerasi, E.; Kaiser, N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 1999, 48, 738–744. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Ehses, J.A.; Böni-Schnetzler, M.; Faulenbach, M.; Donath, M.Y. Macrophages, cytokines and β-cell death in type 2 diabetes. Biochem. Soc. Trans. 2008, 36, 340–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef]
- Mandrup-Poulsen, T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 1996, 39, 1005–1029. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, A.; Suarez-Pinzon, W.L.; Shi, Y.; Morgan, A.R.; Bleackley, R.C. DNA fragmentation is an early event in cytokine-induced islet beta-cell destruction. Diabetologia 1994, 37, 733–738. [Google Scholar] [CrossRef]
- Iwahashi, H.; Hanafusa, T.; Eguchi, Y.; Nakajima, H.; Miyagawa, J.; Itoh, N.; Tomita, K.; Namba, M.; Kuwajima, M.; Noguchi, T.; et al. Cytokine-induced apoptotic cell death in a mouse pancreatic beta-cell line: Inhibition by Bcl-2. Diabetologia 1996, 39, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Frigerio, F.; Maechler, P. The sensitivity of pancreatic β-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 2008, 36, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Grishko, V.; Rachek, L.; Musiyenko, S.; LeDoux, S.P.; Wilson, G.L. Involvement of mtDNA damage in free fatty acid-induced apoptosis. Free Radic. Biol. Med. 2005, 38, 755–762. [Google Scholar] [CrossRef]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Miani, M.; Cardozo, A.K. Signalling danger: Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 2013, 56, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Rebelato, E.; Abdulkader, F.; Krause, M.; Carpinelli, A.; Curi, R. Reactive oxygen and nitrogen species generation, antioxidant defenses, and β-cell function: A critical role for amino acids. J. Endocrinol. 2012, 214, 11–20. [Google Scholar] [CrossRef]
- Gehrmann, W.; Elsner, M.; Lenzen, S. Role of metabolically generated reactive oxygen species for lipotoxicity in pancreatic β-cells. Diabetes Obes. Metab. 2010, 12, 149–158. [Google Scholar] [CrossRef]
- Lenzen, S. Oxidative stress: The vulnerable β-cell. Biochem. Soc. Trans. 2008, 36, 343–347. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Montane, J.; Cadavez, L.; Novials, A. Stress and the inflammatory process: A major cause of pancreatic cell death in type 2 diabetes. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Toyama, H.; Takada, M.; Tanaka, T.; Suzuki, Y.; Kuroda, Y. Characterization of islet-infiltrating immunocytes after pancreas preservation by two-layer (UW/perfluorochemical) cold storage method. Transpl. Proc. 2003, 35, 1503–1505. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.; Carrero, J.A.; Ferris, S.T.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Takeda, K.; Takiguchi, M.; Gotoh, T.; Matsumoto, M.; Wada, I.; Akira, S.; Araki, E.; Mori, M. Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 10845–10850. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Matsuoka, T.A. Role of pancreatic transcription factors in maintenance of mature β-cell function. Int. J. Mol. Sci. 2015, 16, 6281–6297. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- White, M.G.; Shaw, J.A.; Taylor, R. Type 2 diabetes: The pathologic basis of reversible β-cell dysfunction. Diabetes Care 2016, 39, 2080–2088. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Natali, A.; Bell, P.; Cavallo-Perin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. J. Clin. Invest. 1997, 100, 1166–1173. [Google Scholar] [CrossRef]
- Tricò, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. JCI Insight 2018, 3, e124912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalloyer, F.; Vandewalle, B.; Percevault, F.; Torpier, G.; Kerr-Conte, J.; Oosterveer, M.; Paumelle, R.; Fruchart, J.C.; Kuipers, F.; Pattou, F.; et al. Peroxisome proliferator-activated receptor α improves pancreatic adaptation to insulin resistance in obese mice and reduces lipotoxicity in human islets. Diabetes 2006, 55, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleliunas, R.E.; Aljaadi, A.M.; Laher, I.; Glier, M.B.; Green, T.J.; Murphy, M.; Miller, J.W.; Devlin, A.M. Folic acid supplementation of female mice, with or without vitamin B-12, before and during pregnancy and lactation programs adiposity and vascular health in adult male offspring. J. Nutr. 2016, 146, 688–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusens, B.; Theys, N.; Dumortier, O.; Goosse, K.; Remacle, C. Maternal malnutrition programs the endocrine pancreas in progeny. Am. J. Clin. Nutr. 2011, 94, 1824S–1829S. [Google Scholar] [CrossRef]
- Peters, L.; Posgai, A.; Brusko, T.M. Islet–immune interactions in type 1 diabetes: The nexus of beta cell destruction. Clin. Exp. Immunol. 2019, 198, 326–340. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Rodríguez, M.; Raurell-Vila, H.; Colli, M.L.; Alvelos, M.I.; Subirana-Granés, M.; Juan-Mateu, J.; Norris, R.; Turatsinze, J.V.; Nakayasu, E.S.; Webb-Robertson, B.J.; et al. The impact of proinflammatory cytokines on the β-cell regulatory landscape provides insights into the genetics of type 1 diabetes. Nat. Genet. 2019, 51, 1588–1595. [Google Scholar] [CrossRef]
- Böni-Schnetzler, M.; Ehses, J.A.; Faulenbach, M.; Donath, M.Y. Insulitis in type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 201–204. [Google Scholar] [CrossRef]
- Choudhury, S.; Ghosh, S.; Gupta, P.; Mukherjee, S.; Chattopadhyay, S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic. Res. 2015, 49, 1371–1383. [Google Scholar] [CrossRef]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martinez de Marañon, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerf, M.E. Developmental Programming and Glucolipotoxicity: Insights on Beta Cell Inflammation and Diabetes. Metabolites 2020, 10, 444. https://doi.org/10.3390/metabo10110444
Cerf ME. Developmental Programming and Glucolipotoxicity: Insights on Beta Cell Inflammation and Diabetes. Metabolites. 2020; 10(11):444. https://doi.org/10.3390/metabo10110444
Chicago/Turabian StyleCerf, Marlon E. 2020. "Developmental Programming and Glucolipotoxicity: Insights on Beta Cell Inflammation and Diabetes" Metabolites 10, no. 11: 444. https://doi.org/10.3390/metabo10110444
APA StyleCerf, M. E. (2020). Developmental Programming and Glucolipotoxicity: Insights on Beta Cell Inflammation and Diabetes. Metabolites, 10(11), 444. https://doi.org/10.3390/metabo10110444