Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes


Abstract
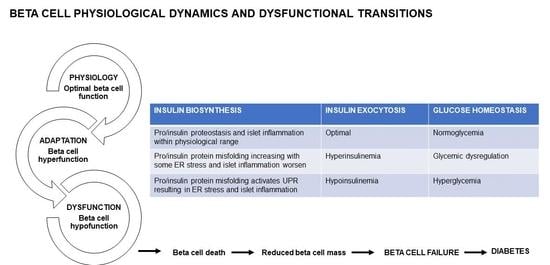
:
{kind=link}
{kind=link}
1. Introduction
2. Systemic and Islet Inflammation in Obesity and Diabetes
3. Mediators of Islet Inflammation
4. Metabolic Interplay of Islet Inflammation, Glucolipotoxicity, and Beta Cell Dysfunction
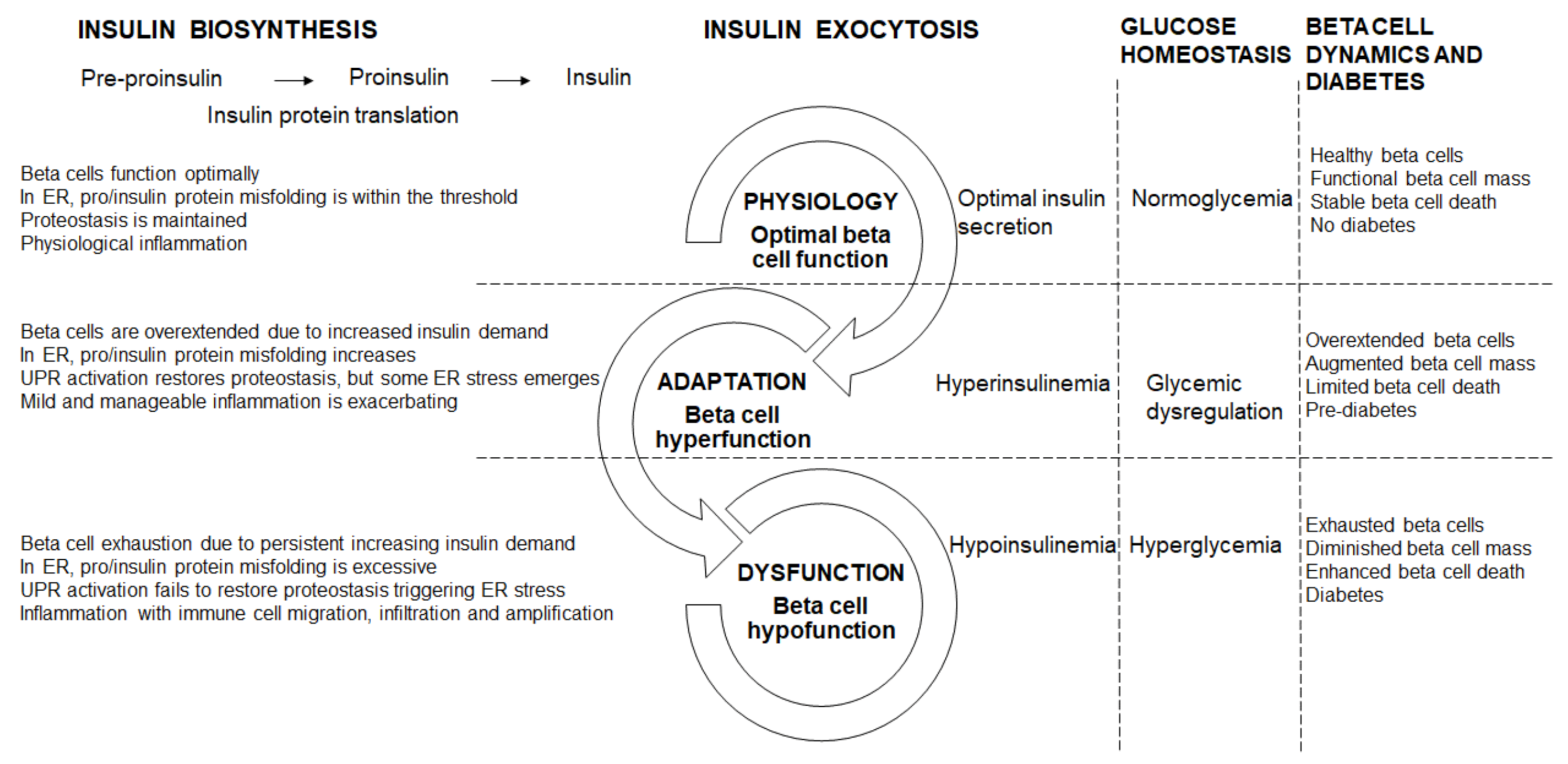
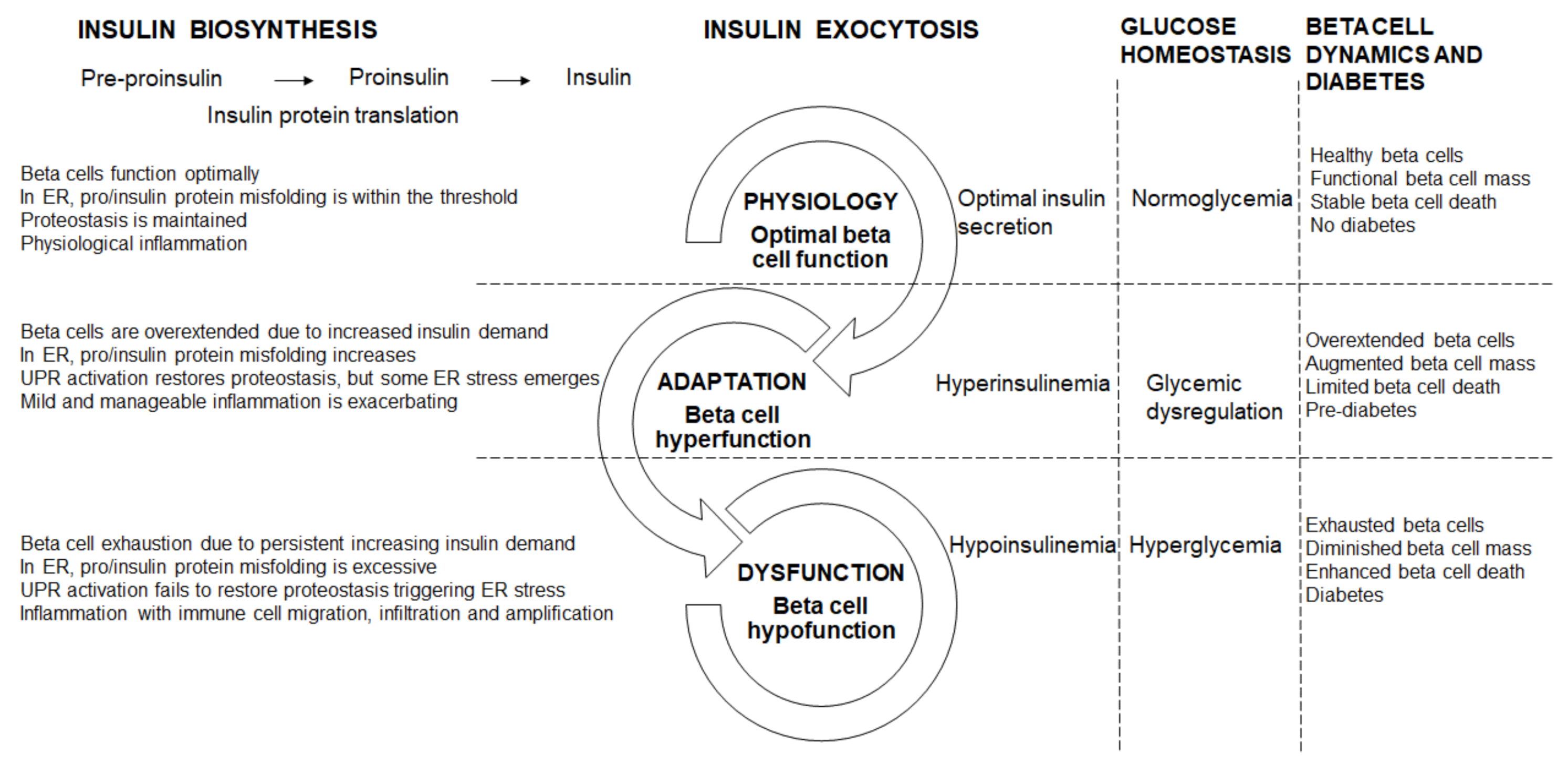
5. Beta Cell Dysfunctional Transitions
5.1. Beta Cell Physiology (Optimal Beta Cell Function)
5.2. Beta Cell Hyperfunction
5.3. Beta Cell Hypofunction
5.4. Beta Cell Failure and Diabetes
6. Conclusions
Funding
Conflicts of Interest
References
- Jeffrey, K.D.; Alejandro, E.U.; Luciani, D.S.; Kalynyak, T.B.; Hu, X.; Li, H.; Lin, Y.; Townsend, R.R.; Polonsky, K.S.; Johnson, J.D. Carboxypeptidase E mediates palmitate-induced β-cell ER stress and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 8452–8457. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Luciani, D.S. Mechanisms of pancreatic β-cell apoptosis in diabetes and its therapies. Adv. Exp. Med. Biol. 2010, 654, 447–462. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Liu, Y.; Hasan, S.D.; Martinez, S.C.; Cras-Méneur, C.; Welling, C.M.; Bernal-Mizrachi, E.; Tanizawa, Y.; Rhodes, C.J.; Zmuda, E.; et al. Glucose and fatty acids synergize to promote β-cell apoptosis through activation of glycogen synthase kinase 3β independent of JNK activation. PLoS ONE 2011, 6, e18146. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and β-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Dobrian, A.D.; Morris, M.A.; Nadler, J.L. Islet inflammation: A unifying target for diabetes treatment? Trends Endocrinol. Metab. 2013, 24, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.E. Adiposopathy: Is ‘sick fat’ a cardiovascular disease? J. Am. Coll. Cardiol. 2011, 57, 2461–2473. [Google Scholar] [CrossRef] [Green Version]
- Carbone, F.; Lattanzio, M.S.; Minetti, S.; Ansaldo, A.M.; Ferrara, D.; Molina-Molina, E.; Belfiore, A.; Elia, E.; Pugliese, S.; Palmieri, V.O.; et al. Circulating CRP levels are associated with epicardial and visceral fat depots in women with metabolic syndrome criteria. Int. J. Mol. Sci. 2019, 20, 5981. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.; Percival, K.; Thelwall, P.E.; Hollingsworth, K.G.; Taylor, R. Altered volume, morphology and composition of the pancreas in type 2 diabetes. PLoS ONE 2015, 10, e0126825. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Victor, R.G.; Mathur, R.; Nelson, M.D.; Szczepaniak, E.W.; Tyer, N.; Chen, I.; Unger, R.H.; Bergman, R.N.; Lingvay, I. Pancreatic steatosis and its relationship to β-cell dysfunction in humans: Racial and ethnic variations. Diabetes Care 2012, 35, 2377–2383. [Google Scholar] [CrossRef] [Green Version]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; Bontemps, S.; Van Waesberghe, J.H.; Schindhelm, R.K.; Mari, A.; Heine, R.J.; Diamant, M. Pancreatic fat content and β-cell function in men with and without type 2 diabetes. Diabetes Care 2007, 30, 2916–2921. [Google Scholar] [CrossRef] [Green Version]
- Van Der Zijl, N.J.; Goossens, G.H.; Moors, C.C.; Van Raalte, D.H.; Muskiet, M.H.; Pouwels, P.J.; Blaak, E.E.; Diamant, M. Ectopic fat storage in the pancreas, liver, and abdominal fat depots: Impact on β-cell function in individuals with impaired glucose metabolism. J. Clin. Endocrinol. Metab. 2011, 96, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Heni, M.; Machann, J.; Staiger, H.; Schwenzer, N.F.; Peter, A.; Schick, F.; Claussen, C.D.; Stefan, N.; Häring, H.U.; Fritsche, A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: A nuclear magnetic resonance study. Diabetes Metab. Res. Rev. 2010, 26, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Böni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Hwang, S.; Lee, S.H.; Lee, Y.R.; Shin, J.; Park, K.S.; Cho, Y.M. Genome-wide identification of palmitate-regulated immediate early genes and target genes in pancreatic beta-cells reveals a central role of NF-κB. Mol. Biol. Rep. 2012, 39, 6781–6789. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Halban, P.A. Decreased beta-cell mass in diabetes: Significance, mechanisms and therapeutic implications. Diabetologia 2004, 47, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Schumann, D.M.; Faulenbach, M.; Ellingsgaard, H.; Perren, A.; Ehses, J.A. Islet inflammation in type 2 diabetes: From metabolic stress to therapy. Diabetes Care 2008, 31, S161–S164. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starling, S. Drivers of islet inflammation. Nat. Rev. Endocrinol. 2019, 15, 128. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourdan, T.; Godlewski, G.; Cinar, R.; Bertola, A.; Szanda, G.; Liu, J.; Tam, J.; Han, T.; Mukhopadhyay, B.; Skarulis, M.C.; et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 2013, 19, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Islet-associated macrophages in type 2 diabetes. Diabetologia 2009, 52, 1686–1688. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Martin, T.M.; Mendoza, T.; Stadler, K.; Alami, W.; Karlstad, M.D.; Robson, M.J.; Blakely, R.D.; et al. Pancreatic deletion of the interleukin-1 receptor disrupts whole body glucose homeostasis and promotes islet β-cell de-differentiation. Mol. Metab. 2018, 14, 95–107. [Google Scholar] [CrossRef]
- Herder, C.; Dalmas, E.; Böni-Schnetzler, M.; Donath, M.Y. The IL-1 pathway in type 2 diabetes and cardiovascular complications. Trends Endocrinol. Metab. 2015, 26, 551–563. [Google Scholar] [CrossRef]
- Kammoun, H.L.; Allen, T.L.; Henstridge, D.C.; Barre, S.; Coll, R.C.; Lancaster, G.I.; Cron, L.; Reibe, S.; Chan, J.Y.; Bensellam, M.; et al. Evidence against a role for NLRP3-driven islet inflammation in db/db mice. Mol. Metab. 2018, 10, 66–73. [Google Scholar] [CrossRef]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, M.; Sahraoui, A.; Høyem, M.; Øgaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. Nlrp3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.; Dai, D.L.; Soukhatcheva, G.; Potter, K.J.; Van Rooijen, N.; Ehses, J.A.; Verchere, C.B. IL-1 blockade attenuates islet amyloid polypeptide-induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J. Immunol. 2011, 187, 2755–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.C.; Courtade, J.A.; Ehses, J.A.; Verchere, C.B. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2015, 58, 575–585. [Google Scholar] [CrossRef]
- Li, Y.; Ding, X.; Fan, P.; Guo, J.; Tian, X.; Feng, X.; Zheng, J.; Tian, P.; Ding, C.; Xue, W. Inactivation of p27kip1 promoted nonspecific inflammation by enhancing macrophage proliferation in islet transplantation. Endocrinology 2016, 157, 4121–4132. [Google Scholar] [CrossRef] [Green Version]
- Annicotte, J.S.; Blanchet, E.; Chavey, C.; Iankova, I.; Costes, S.; Assou, S.; Teyssier, J.; Dalle, S.; Sardet, C.; Fajas, L. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat. Cell Biol. 2009, 11, 1017–1023. [Google Scholar] [CrossRef]
- Grouwels, G.; Cai, Y.; Hoebeke, I.; Leuckx, G.; Heremans, Y.; Ziebold, U.; Stangé, G.; Chintinne, M.; Ling, Z.; Pipeleers, D.; et al. Ectopic expression of E2F1 stimulates β-cell proliferation and function. Diabetes 2010, 59, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Böni-Schnetzler, M.; Häuselmann, S.P.; Dalmas, E.; Meier, D.T.; Thienel, C.; Traub, S.; Schulze, F.; Steiger, L.; Dror, E.; Martin, P.; et al. β cell-specific deletion of the IL-1 receptor antagonist impairs β cell proliferation and insulin secretion. Cell Rep. 2018, 22, 1774–1786. [Google Scholar] [CrossRef] [Green Version]
- Osmai, M.; Osmai, Y.; Bang-Berthelsen, C.H.; Pallesen, E.M.; Vestergaard, A.L.; Novotny, G.W.; Pociot, F.; Mandrup-Poulsen, T. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes Metab. Res. 2016, 32, 334–349. [Google Scholar] [CrossRef] [Green Version]
- Roggli, E.; Britan, A.; Gattesco, S.; Lin-Marq, N.; Abderrahmani, A.; Meda, P.; Regazzi, R. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic β-cells. Diabetes 2010, 59, 978–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, F.A.; Sebastiani, G.; Juan-Mateu, J.; Villate, O.; Marroqui, L.; Ladrière, L.; Tugay, K.; Regazzi, R.; Bugliani, M.; Marchetti, P.; et al. MicroRNAs miR-23a-3p, miR-23b-3p, and miR-149-5p regulate the expression of proapoptotic bh3-only proteins DP5 and PUMA in human pancreatic β-cells. Diabetes 2017, 66, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fred, R.G.; Bang-Berthelsen, C.H.; Mandrup-Poulsen, T.; Grunnet, L.G.; Welsh, N. High glucose suppresses human islet insulin biosynthesis by inducing miR-133a leading to decreased polypyrimidine tract binding protein expression. PLoS ONE 2010, 5, e10843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, A.L.; Bang-Berthelsen, C.H.; Fløyel, T.; Stahl, J.L.; Christen, L.; Sotudeh, F.T.; De Hemmer Horskjñr, P.; Frederiksen, K.S.; Kofod, F.G.; Bruun, C.; et al. MicroRNAs and histone deacetylase inhibition-mediated protection against inflammatory β-cell damage. PLoS ONE 2018, 13, e0203713. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- Cameron, J.E.; Yin, Q.; Fewell, C.; Lacey, M.; McBride, J.; Wang, X.; Lin, Z.; Schaefer, B.C.; Flemington, E.K. Epstein-Barr virus latent membrane protein 1 induces cellular microRNA miR-146a, a modulator of lymphocyte signaling pathways. J. Virol. 2008, 82, 1946–1958. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Zhao, L.; Jin, W.; Xiao, Q.; Peng, Y.; Huang, G.; Li, X.; DaSilva-Arnold, S.; Yu, H.; Zhou, Z. MicroRNA-203a regulates pancreatic β cell proliferation and apoptosis by targeting IRS2. Mol. Biol. Rep. 2020. [Google Scholar] [CrossRef]
- Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [Green Version]
- Laybutt, D.R.; Preston, A.M.; Åkerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Cervin, C.; Lyssenko, V.; Bakhtadze, E.; Lindholm, E.; Nilsson, P.; Tuomi, T.; Cilio, C.M.; Groop, L. Genetic similarities between latent autoimmune diabetes in adults, type 1 diabetes, and type 2 diabetes. Diabetes 2008, 57, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, P1084–P1094. [Google Scholar] [CrossRef]
- Hjort, R.; Ahlqvist, E.; Carlsson, P.O.; Grill, V.; Groop, L.; Martinell, M.; Rasouli, B.; Rosengren, A.; Tuomi, T.; Åsvold, B.O.; et al. Overweight, obesity and the risk of LADA: Results from a Swedish case–control study and the Norwegian HUNT study. Diabetologia 2018, 61, 1333–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharroubi, I.; Ladrière, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: Role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitz, J.R.; Makhmutova, M.; Almaça, J.; Stertmann, J.; Aamodt, K.; Brissova, M.; Speier, S.; Rodriguez-Diaz, R.; Caicedo, A. Mouse pancreatic islet macrophages use locally released ATP to monitor beta cell activity. Diabetologia 2018, 61, 182–192. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Murthy Madiraju, S.R. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef]
- Schrimpe-Rutledge, A.C.; Fontès, G.; Gritsenko, M.A.; Norbeck, A.D.; Anderson, D.J.; Waters, K.M.; Adkins, J.N.; Smith, R.D.; Poitout, V.; Metz, T.O. Discovery of novel glucose-regulated proteins in isolated human pancreatic islets using LC-MS/MS-based proteomics. J. Proteome Res. 2012, 11, 3520–3532. [Google Scholar] [CrossRef]
- Henquin, J.C.; Dufrane, D.; Nenquin, M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes 2006, 55, 3470–3477. [Google Scholar] [CrossRef] [Green Version]
- Schuit, F.; Flamez, D.; De Vos, A.; Pipeleers, D. Glucose-regulated gene expression maintaining the glucose-responsive state of β-cells. Diabetes 2002, 51, S326–S332. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef]
- Schultheis, J.; Beckmann, D.; Mulac, D.; Müller, L.; Esselen, M.; Düfer, M. Nrf2 activation protects mouse beta cells from glucolipotoxicity by restoring mitochondrial function and physiological redox balance. Oxid. Med. Cell. Longev. 2019, 2019, 7518510. [Google Scholar] [CrossRef]
- Hajmrle, C.; Smith, N.; Spigelman, A.F.; Dai, X.; Senior, L.; Bautista, A.; Ferdaoussi, M.; MacDonald, P.E. Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 2016, 1, e86055. [Google Scholar] [CrossRef]
- Henquin, J.C.; Ishiyama, N.; Nenquin, M.; Ravier, M.A.; Jonas, J.C. Signals and pools underlying biphasic insulin secretion. Diabetes 2002, 5, S60–S67. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Cerf, M.E. High fat programming of beta cell compensation, exhaustion, death and dysfunction. Pediatr. Diabetes 2015, 16, 71–78. [Google Scholar] [CrossRef]
- Mezza, T.; Muscogiuri, G.; Sorice, G.P.; Clemente, G.; Hu, J.; Pontecorvi, A.; Holst, J.J.; Giaccari, A.; Kulkarni, R.N. Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 2014, 63, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, S.; Uno, S.; Iwahashi, H.; Fujita, Y.; Yoshikawa, A.; Kozawa, J.; Okita, K.; Takiuchi, D.; Eguchi, H.; Nagano, H.; et al. Predominance of β-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 2053–2061. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Karlstad, M.D.; Regal, K.M.; Sparer, T.E.; Lu, D.; Elks, C.M.; Grant, R.W.; Stephens, J.M.; Burk, D.H.; Collier, J.J. CCL20 is elevated during obesity and differentially regulated by NF-κB subunits in pancreatic β-cells. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 637–652. [Google Scholar] [CrossRef] [Green Version]
- Burke, S.J.; Stadler, K.; Lu, D.; Gleason, E.; Han, A.; Donohoe, D.R.; Rogers, R.C.; Hermann, G.E.; Karlstad, M.D.; Collier, J.J. IL-1β reciprocally regulates chemokine and insulin secretion in pancreatic β-cells via NF-κB. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E715–E726. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [Green Version]
- White, S.A.; Zhang, L.S.; Pasula, D.J.; Yang, Y.H.; Luciani, D.S. Bax and Bak jointly control survival and dampen the early unfolded protein response in pancreatic β-cells under glucolipotoxic stress. Sci. Rep. 2020, 10, 10986. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; Macgregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Zong, W.X.; Li, C.; Hatzivassiliou, G.; Lindsten, T.; Yu, Q.C.; Yuan, J.; Thompson, C.B. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J. Cell Biol. 2003, 162, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Gurzov, E.N.; Germano, C.M.; Cunha, D.A.; Ortis, F.; Vanderwinden, J.M.; Marchetti, P.; Zhang, L.; Eizirik, D.L. p53 up-regulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 19910–19920. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ding, Y.; Zhang, Z.; Dai, X.; Jiang, Y.; Bao, L.; Li, Y. Grape seed proanthocyanidins ameliorate pancreatic beta-cell dysfunction and death in low-dose streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats partially by regulating endoplasmic reticulum stress. Nutr. Metab. 2013, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Novotny, G.W.; Lundh, M.; Backe, M.B.; Christensen, D.P.; Hansen, J.B.; Dahllöf, M.S.; Pallesen, E.M.; Mandrup-Poulsen, T. Transcriptional and translational regulation of cytokine signaling in inflammatory β-cell dysfunction and apoptosis. Arch. Biochem. Biophys. 2012, 528, 171–184. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerf, M.E. High fat programming of beta-cell failure. Adv. Exp. Med. Biol. 2010, 654, 77–89. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerf, M.E. Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes. Metabolites 2020, 10, 452. https://doi.org/10.3390/metabo10110452
Cerf ME. Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes. Metabolites. 2020; 10(11):452. https://doi.org/10.3390/metabo10110452
Chicago/Turabian StyleCerf, Marlon E. 2020. "Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes" Metabolites 10, no. 11: 452. https://doi.org/10.3390/metabo10110452
APA StyleCerf, M. E. (2020). Beta Cell Physiological Dynamics and Dysfunctional Transitions in Response to Islet Inflammation in Obesity and Diabetes. Metabolites, 10(11), 452. https://doi.org/10.3390/metabo10110452