Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics

 ,
, 
 , ,
, ,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
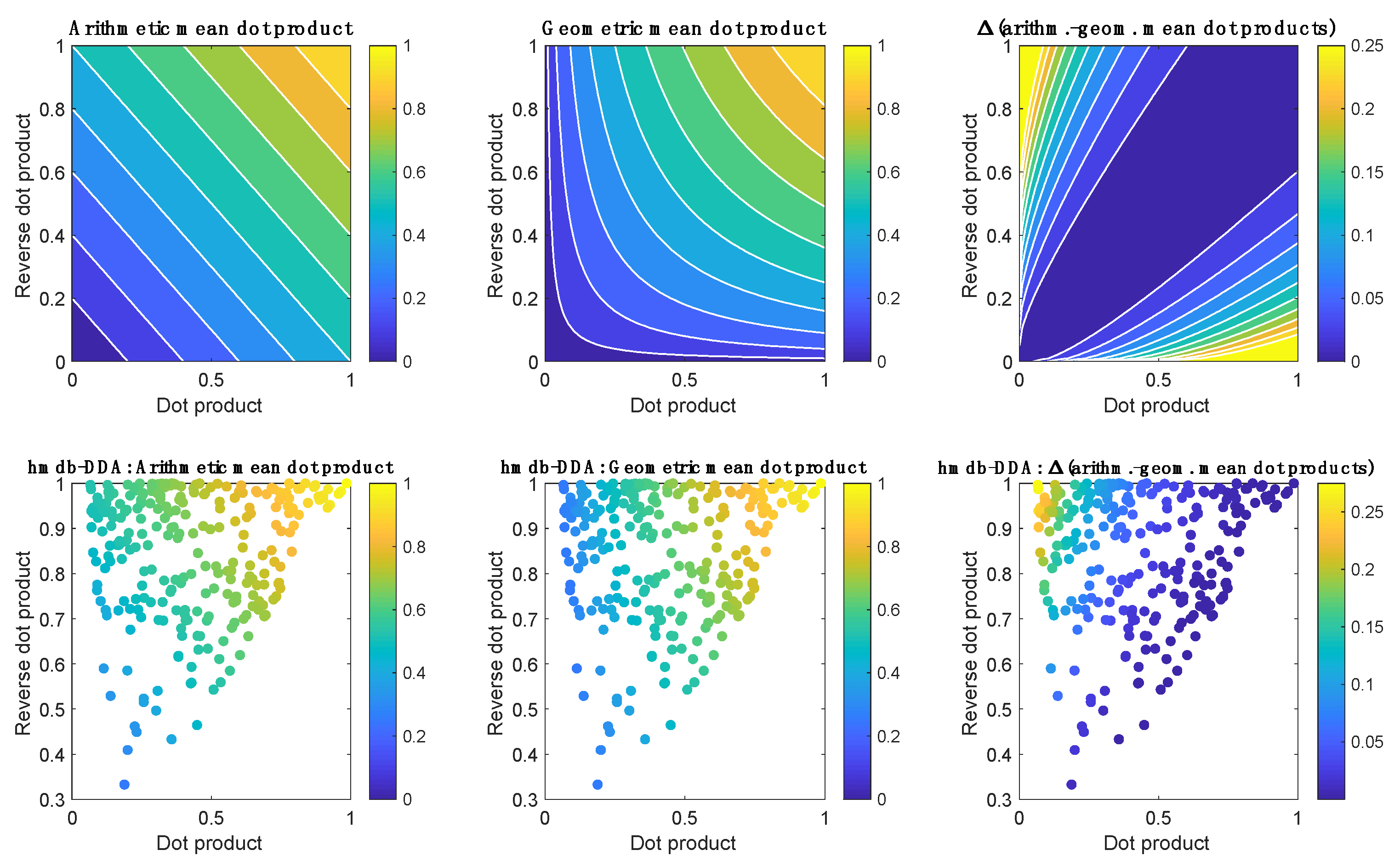
2. Results and Discussion
2.1. Data Overview
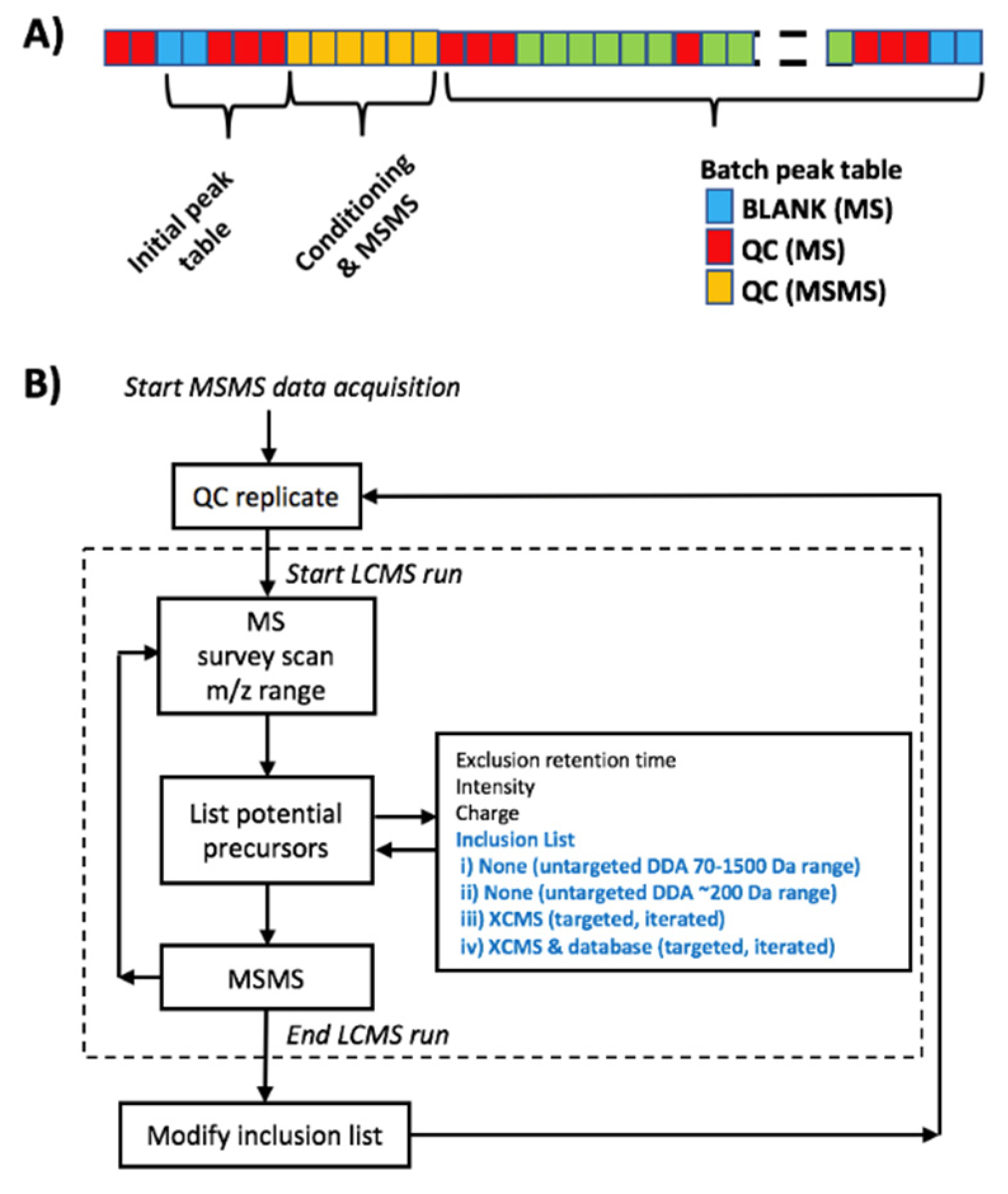
2.2. MS2 Data Dependent Acquisition Strategies
2.3. MS2 Data Dependent Acquisition Strategies in QA/QC Pipelines
3. Materials and Methods
3.1. Standards and Reagents
3.2. Research Ethics
3.3. Sample Preparation
3.4. Sample Analysis
3.5. MS2 Data Dependent Acquisition Methods
3.6. Peak Table Generation and Metabolite Annotation
3.7. Software and Data
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metab. Off. J. Metab. Soc. 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanisevic, J.; Want, E.J. From samples to insights into metabolism: Uncovering biologically relevant information in LC-HRMS metabolomics data. Metabolites 2019, 9, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, G.; Allwood, J.W.; Weber, R.; Brown, M.; Begley, P.; Hollywood, K.A.; Jones, M.; Unwin, R.D.; Bishop, P.N.; Cooper, G.J.S.; et al. A new strategy for MS/MS data acquisition applying multiple data dependent experiments on Orbitrap mass spectrometers in non-targeted metabolomic applications. Metabolomics 2015, 11, 1068–1080. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, R.; He, C.; Su, H.; Ma, H.; Wan, J.-B. An integrated strategy to improve data acquisition and metabolite identification by time-staggered ion lists in UHPLC/Q-TOF MS-based metabolomics. J. Pharm. Biomed. Anal. 2018, 157, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. Sirius 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Considine, E.C.; Thomas, G.; Boulesteix, A.L.; Khashan, A.S.; Kenny, L.C. Critical review of reporting of the data analysis step in metabolomics. Metabolomics 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Villaseñor, A.; Garcia-Perez, I.; Garcia, A.; Posma, J.M.; Fernández-López, M.; Nicholas, A.J.; Modi, N.; Holmes, E.; Barbas, C. Breast milk metabolome characterization in a single-phase extraction, multiplatform analytical approach. Anal. Chem. 2014, 86, 8245–8252. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sena, T.; Luongo, G.; Sanjuan-Herráez, D.; Castell, J.V.; Vento, M.; Quintás, G.; Kuligowski, J. Monitoring of system conditioning after blank injections in untargeted UPLC-MS metabolomic analysis. Sci. Rep. 2019, 9, 9822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuligowski, J.; Sánchez-Illana, Á.; Sanjuán-Herráez, D.; Vento, M.; Quintás, G. Intra-batch effect correction in liquid chromatography-mass spectrometry using quality control samples and support vector regression (QC-SVRC). Analyst 2015, 140, 7810–7817. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Illana, Á.; Pérez-Guaita, D.; Cuesta-García, D.; Sanjuan-Herráez, J.D.; Vento, M.; Ruiz-Cerdá, J.L.; Quintás, G.; Kuligowski, J. Model selection for within-batch effect correction in UPLC-MS metabolomics using quality control—Support vector regression. Anal. Chim. Acta 2018, 1026, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Liu, K.-H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. Camera: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. LipiDex: An Integrated Software Package for High-Confidence Lipid Identification. Cell Syst. 2018, 6, 621–625.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ten-Doménech, I.; Martínez-Sena, T.; Moreno-Torres, M.; Sanjuan-Herráez, J.D.; Castell, J.V.; Parra-Llorca, A.; Vento, M.; Quintás, G.; Kuligowski, J. Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics. Metabolites 2020, 10, 126. https://doi.org/10.3390/metabo10040126
Ten-Doménech I, Martínez-Sena T, Moreno-Torres M, Sanjuan-Herráez JD, Castell JV, Parra-Llorca A, Vento M, Quintás G, Kuligowski J. Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics. Metabolites. 2020; 10(4):126. https://doi.org/10.3390/metabo10040126
Chicago/Turabian StyleTen-Doménech, Isabel, Teresa Martínez-Sena, Marta Moreno-Torres, Juan Daniel Sanjuan-Herráez, José V. Castell, Anna Parra-Llorca, Máximo Vento, Guillermo Quintás, and Julia Kuligowski. 2020. "Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics" Metabolites 10, no. 4: 126. https://doi.org/10.3390/metabo10040126
APA StyleTen-Doménech, I., Martínez-Sena, T., Moreno-Torres, M., Sanjuan-Herráez, J. D., Castell, J. V., Parra-Llorca, A., Vento, M., Quintás, G., & Kuligowski, J. (2020). Comparing Targeted vs. Untargeted MS2 Data-Dependent Acquisition for Peak Annotation in LC–MS Metabolomics. Metabolites, 10(4), 126. https://doi.org/10.3390/metabo10040126