The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
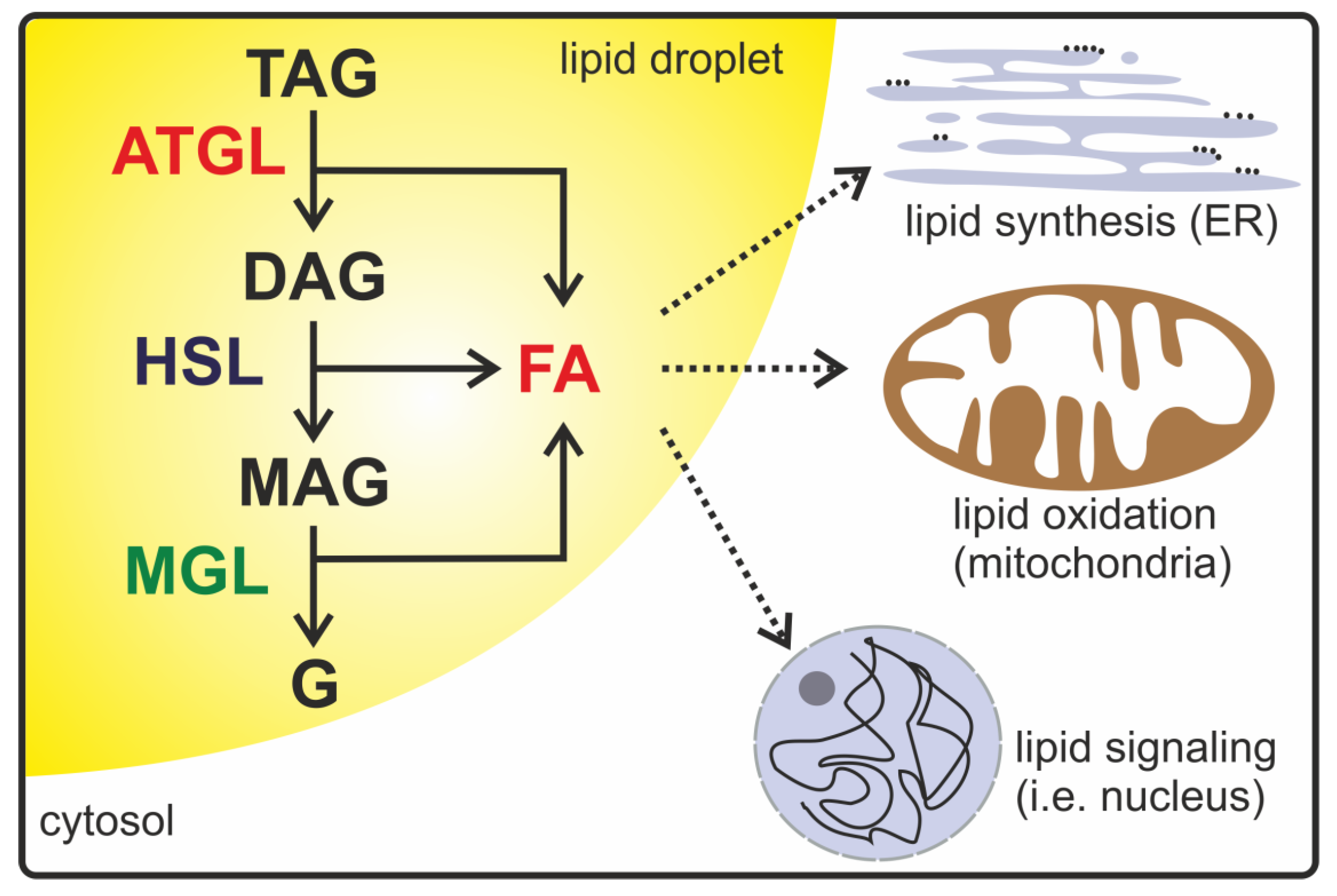
1. Introduction
2. ATGL and Its Interaction Partners
2.1. Alpha/Beta-Hydrolase Domain Containing Protein 5 (ABHD5)
2.2. Perilipins (PLINs)
2.3. G0/G1 Switch Gene 2 (G0S2)
2.4. Hypoxia-Inducible LD-Associated Protein (HILPDA)
2.5. Fat-Specific Protein-27 (FSP-27)
2.6. Pigment Epithelium-Derived Factor (PEDF)
2.7. Ubiquitin Regulatory X Domain-Containing Protein 8 (UBXD8)
2.8. Golgi Brefeldin a Resistance Factor 1 (GBF1)
2.9. 14-3-3
2.10. Peroxisome Biogenesis Factor 5 (PEX5)
3. HSL and Its Interaction Partners
3.1. FABPs
3.2. PLINs
3.3. Vimentin (VIM)
3.4. Cavin-1
3.5. Carbohydrate Response Element Binding Protein (ChREBP)
3.6. Microtubule-Associated Proteins 1A/1B Light Chain 3B (LC3)
3.7. Steroidogenic Acute Regulatory Protein (StAR)
4. MGL and Its Interaction Partners
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoyl glycerol |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| ABHD5 | alpha/beta-hydrolase domain containing protein 5 |
| ADP | adenosine-diphosphate |
| ADRP | adipose differentiation related protein |
| ARF-1 | ADP-ribosylation factor 1 |
| AT | adipose tissue |
| ATGL | adipose triglyceride lipase |
| ATP | adenosine-triphosphate |
| Bcl2 | B-Cell CLL/Lymphoma 2 |
| cAMP | cyclic adenosine monophosphate |
| CBRs | cannabinoid receptors |
| CE | cholesteryl ester |
| CGI-58 | comparative gene identification-58 |
| CHGB | chromogranin B |
| ChREBP | carbohydrate response element binding protein |
| CIDEC | cell death-inducing DNA fragment factor 40/45-like effector C |
| CL 316,243 | β3 adrenoceptor agonist |
| COPI | coatomer protein I |
| DAG | diacylglycerol |
| DNL | de novo lipogenesis |
| ELOVL6; | elongase of long chain fatty acids family 6 |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| FA | fatty acid |
| FABP | fatty acid-binding protein |
| FSP-27 | fat-specific protein-27 |
| G0S2 | G0/G1 switch gene 2 |
| GBF1 | Golgi Brefeldin A resistance factor 1 |
| GH | growth hormone |
| HIG-2 | hypoxia-inducible gene-2 |
| HILPDA | hypoxia-inducible LD-associated protein |
| HSL | hormone-sensitive lipase |
| IC50 | half maximal inhibitory concentration |
| iPLA2-zeta | Ca2+-independent phospholipase A2-zeta |
| KIFC3 | Kinesin Family Member C3 |
| KO | knockout |
| LC3 | microtubule-associated proteins 1A/1B light chain 3B |
| LC3-interacting region | LC3-interacting region |
| LD | lipid droplet |
| MAG | monoacylglycerol |
| MGL | MAG lipase |
| NCL | nucleolin |
| NLSD-M/-I | neutral lipid storage disease with myopathy/with ichthyosis |
| p97/VCP | partner p97 subunit/valosin containing protein |
| PAT | perilipin/ADRP/TIP47 |
| PEDF | pigment epithelium-derived factor |
| PEX5 | peroxisome biogenesis factor 5 |
| PKA | protein kinase A |
| PLIN | perilipin |
| PNPLA | patatin-like phospholipase domain containing |
| PPAR | peroxisome proliferator-activated receptor |
| PTRF | polymerase I and transcript release factor |
| PTS1R | peroxisome target sequence 1 receptor protein |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| SND1 | Staphylococcal nuclease and tudor domain containing 1 |
| StAR | steroidogenic acute regulatory protein |
| TAG | triacylglycerol |
| TIP-47 | tail-interacting protein of 47 kDa |
| UBXD8 | ubiquitin regulatory X domain-containing protein 8 |
| VIM | Vimentin |
References
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Papackova, Z.; Cahova, M. Fatty Acid Signaling: The New Function of Intracellular Lipases. Int. J. Mol. Sci. 2015, 16, 3831–3855. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Xie, H.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 880–899. [Google Scholar] [CrossRef]
- Radner, F.P.W.; Streith, I.E.; Schoiswohl, G.; Schweiger, M.; Kumari, M.; Eichmann, T.O.; Rechberger, G.; Koefeler, H.C.; Eder, S.; Schauer, S.; et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J. Biol. Chem. 2010, 285, 7300–7311. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, X.; Heckmann, B.L.; Saarinen, A.M.; Czyzyk, T.A.; Liu, J. Targeted disruption of G0/G1 switch gene 2 enhances adipose lipolysis, alters hepatic energy balance, and alleviates high-fat diet-induced liver steatosis. Diabetes 2014, 63, 934–946. [Google Scholar] [CrossRef]
- El-Assaad, W.; El-Kouhen, K.; Mohammad, A.H.; Yang, J.; Morita, M.; Gamache, I.; Mamer, O.; Avizonis, D.; Hermance, N.; Kersten, S.; et al. Deletion of the gene encoding G0/G1 switch protein 2 (G0s2) alleviates high-fat-diet-induced weight gain and insulin resistance, and promotes browning of white adipose tissue in mice. Diabetologia 2015, 58, 149–157. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Puri, V.; Murano, I.; Saudek, V.; Semple, R.K.; Dash, S.; Hyden, C.S.S.; Bottomley, W.; Vigouroux, C.; Magré, J.; et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 2009, 1, 280–287. [Google Scholar] [CrossRef]
- Nishino, N.; Tamori, Y.; Tateya, S.; Kawaguchi, T.; Shibakusa, T.; Mizunoya, W.; Inoue, K.; Kitazawa, R.; Kitazawa, S.; Matsuki, Y.; et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Investig. 2008, 118, 2808–2821. [Google Scholar] [CrossRef]
- Egloff, M.-P.; Sarda, L.; Verger, R.; Cambillau, C.; Tilbeurgh, H. Van Crystallographic study of the structure of colipase and of the interaction with pancreatic lipase. Protein Sci. 1995, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A. It takes a village: Channeling fatty acid metabolism and triacylglycerol formation via protein interactomes. J. Lipid Res. 2019, 60, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.-H.; Sul, H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457. [Google Scholar] [CrossRef]
- Notari, L.; Baladron, V.; Aroca-Aguilar, J.D.; Balko, N.; Heredia, R.; Meyer, C.; Notario, P.M.; Saravanamuthu, S.; Nueda, M.-L.; Sanchez-Sanchez, F.; et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem. 2006, 281, 38022–38037. [Google Scholar] [CrossRef]
- Taschler, U.; Schreiber, R.; Chitraju, C.; Grabner, G.F.; Romauch, M.; Wolinski, H.; Haemmerle, G.; Breinbauer, R.; Zechner, R.; Lass, A.; et al. Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 937–945. [Google Scholar] [CrossRef]
- Cornaciu, I.; Boeszoermenyi, A.; Lindermuth, H.; Nagy, H.M.; Cerk, I.K.; Ebner, C.; Salzburger, B.; Gruber, A.; Schweiger, M.; Zechner, R.; et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS ONE 2011, 6, e26349. [Google Scholar] [CrossRef]
- Schweiger, M.; Schoiswohl, G.; Lass, A.; Radner, F.P.W.; Haemmerle, G.; Malli, R.; Graier, W.; Cornaciu, I.; Oberer, M.; Salvayre, R.; et al. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 2008, 283, 17211–17220. [Google Scholar] [CrossRef]
- Xie, X.; Langlais, P.; Zhang, X.; Heckmann, B.L.; Saarinen, A.M.; Mandarino, L.J.; Liu, J. Identification of a novel phosphorylation site in adipose triglyceride lipase as a regulator of lipid droplet localization. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1449–E1459. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.-Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Diwoky, C.; Schoiswohl, G.; Feiler, U.; Wongsiriroj, N.; Abdellatif, M.; Kolb, D.; Hoeks, J.; Kershaw, E.E.; Sedej, S.; et al. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 2017, 26, 753–763.e7. [Google Scholar] [CrossRef]
- Schreiber, R.; Hofer, P.; Taschler, U.; Voshol, P.J.; Rechberger, G.N.; Kotzbeck, P.; Jaeger, D.; Preiss-Landl, K.; Lord, C.C.; Brown, J.M.; et al. Hypophagia and metabolic adaptations in mice with defective ATGL-mediated lipolysis cause resistance to HFD-induced obesity. Proc. Natl. Acad. Sci. USA 2015, 112, 13850–13855. [Google Scholar] [CrossRef]
- Fischer, J.; Lefèvre, C.; Morava, E.; Mussini, J.-M.; Laforêt, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef]
- Subramanian, V.; Rotlienberg, A.; Gomez, C.; Cohen, A.W.; Garcia, A.; Bhattacharyya, S.; Shapiro, L.; Dolios, G.; Wang, R.; Lisanti, M.P.; et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 42062–42071. [Google Scholar] [CrossRef]
- Gruber, A.; Cornaciu, I.; Lass, A.; Schweiger, M.; Poeschl, M.; Eder, C.; Kumari, M.; Schoiswohl, G.; Wolinski, H.; Kohlwein, S.D.; et al. The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 12289–12298. [Google Scholar] [CrossRef]
- Sanders, M.A.; Zhang, H.; Mladenovic, L.; Tseng, Y.Y.; Granneman, J.G. Molecular Basis of ABHD5 Lipolysis Activation. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Grond, S.; Radner, F.P.W.; Eichmann, T.O.; Kolb, D.; Grabner, G.F.; Wolinski, H.; Gruber, R.; Hofer, P.; Heier, C.; Schauer, S.; et al. Skin Barrier Development Depends on CGI-58 Protein Expression during Late-Stage Keratinocyte Differentiation. J. Investig. Dermatol. 2017, 137, 403–413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lefèvre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Lass, A.; Zimmermann, R.; Eichmann, T.O.; Zechner, R. Neutral lipid storage disease: Genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E289–E296. [Google Scholar] [CrossRef] [PubMed]
- Kien, B.; Grond, S.; Haemmerle, G.; Lass, A.; Eichmann, T.O.; Radner, F.P.W. ABHD5 stimulates PNPLA1-mediated -O-acylceramide biosynthesis essential for a functional skin permeability barrier. J. Lipid Res. 2018, 59, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Nara, A.; Nakamichi, S.; Kihara, A. Molecular mechanism of the ichthyosis pathology of Chanarin–Dorfman syndrome: Stimulation of PNPLA1-catalyzed ω-O-acylceramide production by ABHD5. J. Dermatol. Sci. 2018, 92, 245–253. [Google Scholar] [CrossRef]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic overexpression of PNPLA3 I148M in mouse liver causes hepatic steatosis. J. Clin. Investig. 2012, 122, 4130–4144. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.-M.V.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef]
- Hofer, P.; Boeszoermenyi, A.; Jaeger, D.; Feiler, U.; Arthanari, H.; Mayer, N.; Zehender, F.; Rechberger, G.; Oberer, M.; Zimmermann, R.; et al. Fatty acid-binding proteins interact with comparative gene identification-58 linking lipolysis with lipid ligand shuttling. J. Biol. Chem. 2015, 290, 18438–18453. [Google Scholar] [CrossRef]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2010, 7, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Kimmel, A.R. Perilipins: Lipid droplet coat proteins adapted for tissue-specific energy storage and utilization, and lipid cytoprotection. Biochimie 2014, 96, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Tansey, J.T.; Sztalryd, C.; Gruia-Gray, J.; Roush, D.L.; Zee, J.V.; Gavrilova, O.; Reitman, M.L.; Deng, C.X.; Li, C.; Kimmel, A.R.; et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. USA 2001, 98, 6494–6499. [Google Scholar] [CrossRef]
- Gandotra, S.; Lim, K.; Girousse, A.; Saudek, V.; O’Rahilly, S.; Savage, D.B. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5). J. Biol. Chem. 2011, 286, 34998–35006. [Google Scholar] [CrossRef]
- Gandotra, S.; Le Dour, C.; Bottomley, W.; Cervera, P.; Giral, P.; Reznik, Y.; Charpentier, G.; Auclair, M.; Delépine, M.; Barroso, I.; et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med. 2011, 364, 740–748. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Moos, M.C.; Londos, C.; Kimmel, A.R. Isolation of cDNAs for perilipins A and B: Sequence and expression of lipid droplet-associated proteins of adipocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 12035–12039. [Google Scholar] [CrossRef]
- Sahu-Osen, A.; Montero-Moran, G.; Schittmayer, M.; Fritz, K.; Dinh, A.; Chang, Y.-F.; McMahon, D.; Boeszoermenyi, A.; Cornaciu, I.; Russell, D.; et al. CGI-58/ABHD5 is phosphorylated on Ser239 by protein kinase A: Control of subcellular localization. J. Lipid Res. 2015, 56, 109–121. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.P.H.; Krishnamoorthy, R.; Rathod, M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 2009, 284, 34538–34544. [Google Scholar] [CrossRef]
- Granneman, J.G.; Moore, H.P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with adipose triglyceride lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef]
- Macpherson, R.E.K.; Vandenboom, R.; Roy, B.D.; Peters, S.J. Skeletal muscle PLIN3 and PLIN5 are serine phosphorylated at rest and following lipolysis during adrenergic or contractile stimulation. Physiol. Rep. 2013, 1, e00084. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Omatsu, N.; Matsushita, S.; Osumi, T. CGI-58 interacts with perilipin and is localized to lipid droplets: Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J. Biol. Chem. 2004, 279, 30490–30497. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Yang, W.; Kozusko, K.; Saudek, V.; Savage, D.B. Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 9163–9168. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.H.-J.; Chan, L.; Heird, W.C.; Nannegari, V.; Taniguchi, S.; Paul, A.; Li, L. Protection against Fatty Liver but Normal Adipogenesis in Mice Lacking Adipose Differentiation-Related Protein. Mol. Cell. Biol. 2006, 26, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Sohn, J.H.; Han, J.S.; Park, Y.J.; Jeon, Y.G.; Ji, Y.; Dalen, K.T.; Sztalryd, C.; Kimmel, A.R.; Kim, J.B. Perilipin 3 deficiency stimulates thermogenic Beige adipocytes through PPARa activation. Diabetes 2018, 67, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Wang, H.; Chen, H.; McLenithan, J.C.; Gong, D.W.; Yang, R.Z.; Yu, D.; Fried, S.K.; Quon, M.J.; Londos, C.; et al. Consequences of lipid droplet coat protein downregulation in liver cells: Abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes 2008, 57, 2037–2045. [Google Scholar] [CrossRef]
- Wolins, N.E.; Skinner, J.R.; Schoenfish, M.J.; Tzekov, A.; Bensch, K.G.; Bickel, P.E. Adipocyte protein S3-12 coats nascent lipid droplets. J. Biol. Chem. 2003, 278, 37713–37721. [Google Scholar] [CrossRef]
- Čopič, A.; Antoine-Bally, S.; Giménez-Andrés, M.; La Torre Garay, C.; Antonny, B.; Manni, M.M.; Pagnotta, S.; Guihot, J.; Jackson, C.L. A giant amphipathic helix from a perilipin that is adapted for coating lipid droplets. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Wu, X.; Chan, L.; Chen, W.; Li, L.; Sleeman, M.; Chang, B. Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am. J. Physiol. Metab. 2013, 304, E770–E779. [Google Scholar]
- Wu, J.; Li, Z.; Hu, P.; Liu, F.; Zhang, X.; Zhao, Y.; Wang, C.; Zhang, H.; Li, L.; Xu, Y.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2014, 61, 870–882. [Google Scholar]
- Drevinge, C.; Dalen, K.T.; Mannila, M.N.; Täng, M.S.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Mardani, I.; Haugen, F.; Fogelstrand, P.; et al. Perilipin 5 is protective in the ischemic heart. Int. J. Cardiol. 2016, 219, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.R.; Mokhtar, R.; Matzaris, M.; Selathurai, A.; Kowalski, G.M.; Mokbel, N.; Meikle, P.J.; Bruce, C.R.; Watt, M.J. PLIN5 deletion remodels intracellular lipid composition and causes insulin resistance in muscle. Mol. Metab. 2014, 3, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Jaeger, D.; Kolleritsch, S.; Zimmermann, R.; Zechner, R.; Lass, A.; Haemmerle, G. The interplay of protein kinase A and Perilipin 5 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 1295–1306. [Google Scholar] [CrossRef]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef]
- Yang, X.; Lu, X.; Lombès, M.; Rha, G.B.; Chi, Y.-I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef]
- Zandbergen, F.; Mandard, S.; Escher, P.; Tan, N.S.; Patsouris, D.; Jatkoe, T.; Rojas-Caro, S.; Madore, S.; Wahli, W.; Tafuri, S.; et al. The G0/G1 switch gene 2 is a novel PPAR target gene. Biochem. J. 2005, 392, 313–324. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Zhang, X.; Xie, X.; Saarinen, A.; Lu, X.; Yang, X.; Liu, J. Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 switch gene 2 (G0S2). J. Biol. Chem. 2014, 289, 1905–1916. [Google Scholar] [CrossRef]
- Heier, C.; Radner, F.P.W.; Moustafa, T.; Schreiber, R.; Grond, S.; Eichmann, T.O.; Schweiger, M.; Schmidt, A.; Cerk, I.K.; Oberer, M.; et al. G0/G1 switch gene 2 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 26141–26150. [Google Scholar] [CrossRef]
- Kioka, H.; Kato, H.; Fujikawa, M.; Tsukamoto, O.; Suzuki, T.; Imamura, H.; Nakano, A.; Higo, S.; Yamazaki, S.; Matsuzaki, T.; et al. Evaluation of intramitochondrial ATP levels identifies G0/G1 switch gene 2 as a positive regulator of oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 273–278. [Google Scholar] [CrossRef]
- Yamada, T.; Park, C.S.; Burns, A.; Nakada, D.; Lacorazza, H.D. The cytosolic protein G0S2 maintains quiescence in hematopoietic stem cells. PLoS ONE 2012, 7, e38280. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.; Santra, M.K.; El-Assaad, W.; Zhu, X.; Huber, W.E.; Keys, R.A.; Teodoro, J.G.; Green, M.R. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009, 69, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schödel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6, e31132. [Google Scholar] [CrossRef]
- DiStefano, M.T.; Danai, L.V.; Roth Flach, R.J.; Chawla, A.; Pedersen, D.J.; Guilherme, A.; Czech, M.P. The lipid droplet protein hypoxia-inducible gene 2 promotes hepatic triglyceride deposition by inhibiting lipolysis. J. Biol. Chem. 2015, 290, 15175–15184. [Google Scholar] [CrossRef]
- Padmanabha Das, K.M.; Wechselberger, L.; Liziczai, M.; De la Rosa Rodriguez, M.; Grabner, G.F.; Heier, C.; Viertlmayr, R.; Radler, C.; Lichtenegger, J.; Zimmermann, R.; et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J. Lipid Res. 2018, 59, 531–541. [Google Scholar] [CrossRef]
- Dijk, W.; Mattijssen, F.; De La Rosa Rodriguez, M.; Valdes, A.L.; Loft, A.; Mandrup, S.; Kalkhoven, E.; Qi, L.; Borst, J.W.; Kersten, S. Hypoxia-inducible lipid droplet-associated is not a direct physiological regulator of lipolysis in adipose tissue. Endocrinology 2017, 158, 1231–1251. [Google Scholar] [CrossRef]
- Puri, V.; Konda, S.; Ranjit, S.; Aouadi, M.; Chawla, A.; Chouinard, M.; Chakladar, A.; Czech, M.P. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 2007, 282, 34213–34218. [Google Scholar] [CrossRef]
- Gong, J.; Sun, Z.; Wu, L.; Xu, W.; Schieber, N.; Xu, D.; Shui, G.; Yang, H.; Parton, R.G.; Li, P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 2011, 195, 953–963. [Google Scholar] [CrossRef]
- Wang, J.; Yan, C.; Xu, C.; Chua, B.T.; Li, P.; Chen, F.J. Polybasic RKKR motif in the linker region of lipid droplet (LD)–associated protein CIDEC inhibits LD fusion activity by interacting with acidic phospholipids. J. Biol. Chem. 2018, 293, 19330–19343. [Google Scholar] [CrossRef]
- Grahn, T.H.M.; Zhang, Y.; Lee, M.J.; Sommer, A.G.; Mostoslavsky, G.; Fried, S.K.; Greenberg, A.S.; Puri, V. FSP27 and PLIN1 interaction promotes the formation of large lipid droplets in human adipocytes. Biochem. Biophys. Res. Commun. 2013, 432, 296–301. [Google Scholar] [CrossRef]
- Sun, Z.; Gong, J.; Wu, H.; Xu, W.; Wu, L.; Xu, D.; Gao, J.; Wu, J.W.; Yang, H.; Yang, M.; et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 2013, 4, 1594. [Google Scholar] [CrossRef]
- Grahn, T.H.M.; Kaur, R.; Yin, J.; Schweiger, M.; Sharma, V.M.; Lee, M.J.; Ido, Y.; Smas, C.M.; Zechner, R.; Lass, A.; et al. Fat-specific protein 27 (FSP27) interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J. Biol. Chem. 2014, 289, 12029–12039. [Google Scholar] [CrossRef]
- Yang, X.; Heckmann, B.L.; Zhang, X.; Smas, C.M.; Liu, J. Distinct Mechanisms Regulate ATGL-Mediated Adipocyte Lipolysis by Lipid Droplet Coat Proteins. Mol. Endocrinol. 2013, 27, 116–126. [Google Scholar] [CrossRef]
- Sharma, V.M.; Vestergaard, E.T.; Jessen, N.; Kolind-Thomsen, P.; Nellemann, B.; Nielsen, T.S.; Vendelbo, M.H.; Møller, N.; Sharma, R.; Lee, K.Y.; et al. Growth hormone acts along the PPARγ-FSP27 axis to stimulate lipolysis in human adipocytes. Am. J. Physiol.-Endocrinol. Metab. 2019, 316, E34–E42. [Google Scholar] [CrossRef] [PubMed]
- Filleur, S.; Nelius, T.; De Riese, W.; Kennedy, R.C. Characterization of pedf: A multi-functional serpin family protein. J. Cell. Biochem. 2009, 106, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Famulla, S.; Lamers, D.; Hartwig, S.; Passlack, W.; Horrighs, A.; Cramer, A.; Lehr, S.; Sell, H.; Eckel, J. Pigment epithelium-derived factor (PEDF) is one of the most abundant proteins secreted by human adipocytes and induces insulin resistance and inflammatory signaling in muscle and fat cells. Int. J. Obes. 2011, 35, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhou, T.; Li, C.; Qi, W.; Mao, Y.; Lu, J.; Yao, Y.; Li, L.; Zhang, T.; Hong, H.; et al. Intracellular pigment epithelium-derived factor contributes to triglyceride degradation. Int. J. Biochem. Cell Biol. 2013, 45, 2076–2086. [Google Scholar] [CrossRef]
- Chung, C.; Doll, J.A.; Gattu, A.K.; Shugrue, C.; Cornwell, M.; Fitchev, P.; Crawford, S.E. Anti-angiogenic pigment epithelium-derived factor regulates hepatocyte triglyceride content through adipose triglyceride lipase (ATGL). J. Hepatol. 2008, 48, 471–478. [Google Scholar] [CrossRef]
- Borg, M.L.; Andrews, Z.B.; Duh, E.J.; Zechner, R.; Meikle, P.J.; Watt, M.J. Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes 2011, 60, 1458–1466. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, T.; Jiang, X.; Yu, H.; Wang, M.; Wei, T.; Cui, H.; Zhuang, W.; Liu, Z.; Zhang, Z.; et al. PEDF and PEDF-derived peptide 44mer stimulate cardiac triglyceride degradation via ATGL. J. Transl. Med. 2015, 13, 1–12. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Richter, C.M.; Kopito, R.R. Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 1345–1350. [Google Scholar] [CrossRef]
- Imai, N.; Suzuki, M.; Hayashi, K.; Ishigami, M.; Hirooka, Y.; Abe, T.; Shioi, G.; Goto, H.; Fujimoto, T. Hepatocyte-specific depletion of UBXD8 induces periportal steatosis in mice fed a high-fat diet. PLoS ONE 2015, 10, e0127114. [Google Scholar] [CrossRef]
- Ellong, E.N.; Soni, K.G.; Bui, Q.T.; Sougrat, R.; Golinelli-Cohen, M.P.; Jackson, C.L. Interaction between the triglyceride lipase ATGL and the arf1 activator GBF1. PLoS ONE 2011, 6, e21889. [Google Scholar] [CrossRef]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-Directional Protein Transport Between the Er and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Glick, B.S. The Mechanisms of Vesicle Budding and Fusion. Cell 2004, 116, 153–166. [Google Scholar] [CrossRef]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef]
- Beller, M.; Sztalryd, C.; Southall, N.; Bell, M.; Jäckle, H.; Auld, D.S.; Oliver, B. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008, 6, 2530–2549. [Google Scholar] [CrossRef]
- Guo, Y.; Walther, T.C.; Rao, M.; Stuurman, N.; Goshima, G.; Terayama, K.; Wong, J.S.; Vale, R.D.; Walter, P.; Farese, R.V. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 2008, 453, 657–661. [Google Scholar] [CrossRef]
- Kleppe, R.; Martinez, A.; Døskeland, S.O.; Haavik, J. The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 2011, 22, 713–719. [Google Scholar] [CrossRef]
- Takahashi, Y. The 14-3-3 proteins: Gene, gene expression, and function. Neurochem. Res. 2003, 28, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B.; Rittinger, K.; Volinia, S.; Caron, P.R.; Aitken, A.; Leffers, H.; Gamblin, S.J.; Smerdon, S.J.; Cantley, L.C. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 1997, 91, 961–971. [Google Scholar] [CrossRef]
- Xie, M. AMPK-Dependent Regulation of Lipid Metabolism in the C. elegans Dauer Larva. Ph.D. Thesis, McGill University Montreal, Montréal, QC, Canada, 2013. [Google Scholar]
- Gould, S.J.; Collins, C.S. Peroxisomal-protein import: Is it really that complex? Nat. Rev. Mol. Cell Biol. 2002, 3, 382–389. [Google Scholar] [CrossRef]
- Harper, C.C.; Berg, J.M.; Gould, S.J. PEX5 binds the PTS1 independently of Hsp70 and the peroxin PEX12. J. Biol. Chem. 2003, 278, 7897–7901. [Google Scholar] [CrossRef]
- Kong, J.; Ji, Y.; Jeon, Y.G.; Han, J.S.; Han, K.H.; Lee, J.H.; Lee, G.; Jang, H.; Choe, S.S.; Baes, M.; et al. Spatiotemporal contact between peroxisomes and lipid droplets regulates fasting-induced lipolysis via PEX5. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Vaughan, M.; Berger, J.F.; Steinberg, D. Hormone-sensitive Lipase and Monoglyceride Activities in Adipose Tissue. J. Biol. Chem. 1964, 239, 401–409. [Google Scholar]
- Rizack, M.A. Activation of an epinephrine-sensitive lipolytic activity from adipose tissue by adenosine 3′,5′-phosphate. J. Biol. Chem. 1964, 239, 392–395. [Google Scholar]
- Hollenberger, C.H.; Raben, M.S.; Astwood, E.B. The lipolytic response to corticotropin. Endocrinology 1961, 68, 589–598. [Google Scholar] [CrossRef]
- Lampidonis, A.D.; Rogdakis, E.; Voutsinas, G.E.; Stravopodis, D.J. The resurgence of Hormone-Sensitive Lipase (HSL) in mammalian lipolysis. Gene 2011, 477, 1–11. [Google Scholar] [CrossRef]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef]
- Marvyn, P.M.; Bradley, R.M.; Button, E.B.; Mardian, E.B.; Duncan, R.E. Fasting upregulates adipose triglyceride lipase and hormone-sensitive lipase levels and phosphorylation in mouse kidney. Biochem. Cell Biol. 2015, 93, 262–267. [Google Scholar] [CrossRef]
- Fredrikson, G.; Tornqvist, H.; Belfrage, P. Hormone-sensitive lipase and monoacylglycerol lipase are both required for complete degradation of adipocyte triacylglycerol. Biochim. Biophys. Acta 1986, 876, 288–293. [Google Scholar] [CrossRef]
- Wei, S.; Lai, K.; Patel, S.; Piantedosi, R.; Shen, H.; Colantuoni, V.; Kraemer, F.B.; Blaner, W.S. Retinyl ester hydrolysis and retinol efflux from BFC-1 beta adipocytes. J. Biol. Chem. 1997, 272, 14159–14165. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.G.; Yeaman, S.J.; Strålfors, P.; Fredrikson, G.; Belfrage, P. Direct evidence that cholesterol ester hydrolase from adrenal cortex is the same enzyme as hormone-sensitive lipase from adipose tissue. Eur. J. Biochem. 1982, 125, 245–249. [Google Scholar] [CrossRef]
- Lee, F.T.; Adams, J.B.; Garton, A.J.; Yeaman, S.J. Hormone-sensitive lipase is involved in the hydrolysis of lipoidal derivatives of estrogens and other steroid hormones. Biochim. Biophys. Acta (BBA) 1988, 963, 258–264. [Google Scholar] [CrossRef]
- Holm, C.; Osterlund, T. Hormone-sensitive lipase and neutral cholesteryl ester lipase. Methods Mol. Biol. 1999, 109, 109–121. [Google Scholar]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive Lipase Deficiency in Mice Causes Diglyceride Accumulation in Adipose Tissue, Muscle, and Testis. J. Biol. Chem. 2002, 277, 4806–4815. [Google Scholar] [CrossRef]
- Osuga, J.I.; Ishibashi, S.; Oka, T.; Yagyu, H.; Tozawa, R.; Fujimoto, A.; Shionoiri, F.; Yahagi, N.; Kraemer, F.B.; Tsutsumi, O.; et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 787–792. [Google Scholar] [CrossRef]
- Xia, B.; Cai, G.H.; Yang, H.; Wang, S.P.; Mitchell, G.A.; Wu, J.W. Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice. PLoS Genet. 2017, 13, 1–17. [Google Scholar] [CrossRef]
- Albert, J.S.; Yerges-Armstrong, L.M.; Horenstein, R.B.; Pollin, T.I.; Sreenivasan, U.T.; Chai, S.; Blaner, W.S.; Snitker, S.; O’Connell, J.R.; Gong, D.-W.; et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 2014, 370, 2307–2315. [Google Scholar] [CrossRef]
- Shen, W.J.; Patel, S.; Hong, R.; Kraemer, F.B. Hormone-sensitive lipase functions as an oligomer. Biochemistry 2000, 39, 2392–2398. [Google Scholar] [CrossRef]
- Østerlund, T.; Danielsson, B.; Degerman, E.; Contreras, J.A.; Edgren, G.; Davis, R.C.; Schotz, M.C.; Holm, C. Domain-structure analysis of recombinant rat hormone-sensitive lipase. Biochem. J. 1996, 319, 411–420. [Google Scholar] [CrossRef]
- ØSterlund, T.; Beussman, D.J.; Julenius, K.; Poon, P.H.; Linse, S.; Shabanowitz, J.; Hunt, D.F.; Schotz, M.C.; Derewenda, Z.S.; Holm, C. Domain identification of hormone-sensitive lipase by circular dichroism and fluorescence spectroscopy, limited proteolysis, and mass spectrometry. J. Biol. Chem. 1999, 274, 15382–15388. [Google Scholar] [CrossRef]
- Østerlund, T.; Contreras, J.A.; Holm, C. Identification of essential aspartic acid and histidine residues of hormone-sensitive lipase: Apparent residues of the catalytic triad. FEBS Lett. 1997, 403, 259–262. [Google Scholar] [CrossRef]
- Anthonsen, M.W.; Rönnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef]
- Garton, A.J.; Campbell, D.G.; Cohen, P.; Yeaman, S.J. Primary structure of the site on bovine hormone-sensitive lipase phosphorylated by cyclic AMP-dependent protein kinase. FEBS Lett. 1988, 229, 68–72. [Google Scholar] [CrossRef]
- GartonN, A.J.; Yeaman, S.J. Identification and role of the basal phosphorylation site on hormone-sensitive lipase. Eur. J. Biochem. 1990, 191, 245–250. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Shen, W.J.; Muliro, K.; Patel, S.; Souza, S.C.; Roth, R.A.; Kraemer, F.B. Stimulation of Lipolysis and Hormone-sensitive Lipase via the Extracellular Signal-regulated Kinase Pathway. J. Biol. Chem. 2001, 276, 45456–45461. [Google Scholar] [CrossRef]
- Krintel, C.; Osmark, P.; Larsen, M.R.; Resjö, S.; Logan, D.T.; Holm, C. Ser649 and Ser650 are the major determinants of protein kinase A-mediated activation of human hormone-sensitive lipase against lipid substrates. PLoS ONE 2008, 3, e3756. [Google Scholar] [CrossRef]
- Stralfors, P.; Belfrage, P. Phosphorylation of hormone-sensitive lipase by cyclic AMP-dependent protein kinase. J. Biol. Chem. 1983, 258, 15146–15152. [Google Scholar] [PubMed]
- Kraemer, F.B.; Shen, W.J. Hormone-sensitive lipase: Control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid Res. 2002, 43, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sumida, M.; Birchbauer, A.; Schotz, M.C.; Reue, K. Isolation and characterization of the gene for mouse hormone-sensitive lipase. Genomics 1994, 24, 259–265. [Google Scholar] [CrossRef]
- Smith, A.J.; Sanders, M.A.; Thompson, B.R.; Londos, C.; Kraemer, F.B.; Bernlohr, D.A. Physical association between the adipocyte fatty acid-binding protein and hormone-sensitive lipase: A fluorescence resonance energy transfer analysis. J. Biol. Chem. 2004, 279, 52399–52405. [Google Scholar] [CrossRef]
- Shen, W.J.; Sridhar, K.; Bernlohr, D.A.; Kraemer, F.B. Interaction of rat hormone-sensitive lipase with adipocyte lipid-binding protein. Proc. Natl. Acad. Sci. USA 1999, 96, 5528–5532. [Google Scholar] [CrossRef]
- Smith, A.J.; Thompson, B.R.; Sanders, M.A.; Bernlohr, D.A. Interaction of the adipocyte fatty acid-binding protein with the hormone-sensitive lipase: Regulation by fatty acids and phosphorylation. J. Biol. Chem. 2007, 282, 32424–32432. [Google Scholar] [CrossRef]
- Smith, A.J.; Sanders, M.A.; Juhlmann, B.E.; Hertzel, A.V.; Bernlohr, D.A. Mapping of the hormone-sensitive lipase binding site on the adipocyte fatty acid-binding protein (AFABP): Identification of the charge quartet on the AFABP/aP2 helix-turn-helix domain. J. Biol. Chem. 2008, 283, 33536–33543. [Google Scholar] [CrossRef]
- Jenkins-Kruchten, A.E.; Bennaars-Eiden, A.; Ross, J.R.; Shen, W.J.; Kraemer, F.B.; Bernlohr, D.A. Fatty acid-binding protein-hormone-sensitive lipase interaction: Fatty acid dependence on binding. J. Biol. Chem. 2003, 278, 47636–47643. [Google Scholar] [CrossRef]
- Shen, W.J.; Liang, Y.; Hong, R.; Patel, S.; Natu, V.; Sridhar, K.; Jenkins, A.; Bernlohr, D.A.; Kraemer, F.B. Characterization of the Functional Interaction of Adipocyte Lipid-binding Protein with Hormone-sensitive Lipase. J. Biol. Chem. 2001, 276, 49443–49448. [Google Scholar] [CrossRef]
- Coe, N.R.; Simpson, M.A.; Bernlohr, D.A. Targeted disruption of the adipocyte lipid-binding protein (aP2 protein) gene impairs fat cell lipolysis and increases cellular fatty acid levels. J. Lipid Res. 1999, 40, 967–972. [Google Scholar]
- Londos, C.; Brasaemle, D.L.; Schultz, C.J.; Adler-Wailes, D.C.; Levin, D.M.; Kimmel, A.R.; Rondinone, C.M. On the control of lipolysis in adipocytes. Ann. N. Y. Acad. Sci. 1999, 892, 155–168. [Google Scholar] [CrossRef]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Levin, D.M.; Adler-Wailes, D.C.; Londos, C. The lipolytic stimulation of 3T3-L1 adipocytes promotes the translocation of hormone-sensitive lipase to the surfaces of lipid storage droplets. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2000, 1483, 251–262. [Google Scholar] [CrossRef]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef]
- Sztalryd, C.; Xu, G.; Dorward, H.; Tansey, J.T.; Contreras, J.A.; Kimmel, A.R.; Londos, C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 2003, 161, 1093–1103. [Google Scholar] [CrossRef]
- Egan, J.J.; Greenberg, A.S.; Chang, M.K.; Wek, S.A.; Moos, M.C.; Londos, C. Mechanism of hormone-stimulated lipolysis in adipocytes: Translocation of hormone-sensitive lipase to the lipid storage droplet. Proc. Natl. Acad. Sci. USA 1992, 89, 8537–8541. [Google Scholar] [CrossRef]
- Wang, H.; Hu, L.; Dalen, K.; Dorward, H.; Marcinkiewicz, A.; Russell, D.; Gong, D.; Londos, C.; Yamaguchi, T.; Holm, C.; et al. Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins. J. Biol. Chem. 2009, 284, 32116–32125. [Google Scholar] [CrossRef]
- Shen, W.; Patel, S.; Miyoshi, H.; Greenberg, A.S.; Kraemer, F.B.; Endocrinology, D.; Palo, V.A.; Health, A. Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J. Lipid Res. 2009, 50, 2306–2313. [Google Scholar] [CrossRef]
- Garcia, A.; Subramanian, V.; Sekowski, A.; Bhattacharyya, S.; Love, M.W.; Brasaemle, D.L. The Amino and Carboxyl Termini of Perilipin A Facilitate the Storage of Triacylglycerols. J. Biol. Chem. 2004, 279, 8409–8416. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Dolios, G.; Shapiro, L.; Wang, R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 46835–46842. [Google Scholar] [CrossRef]
- Shen, W.J.; Patel, S.; Eriksson, J.E.; Kraemer, F.B. Vimentin is a functional partner of hormone sensitive lipase and facilitates lipolysis. J. Proteome Res. 2010, 9, 1786–1794. [Google Scholar] [CrossRef]
- Kumar, N.; Robidoux, J.; Daniel, K.W.; Guzman, G.; Floering, L.M.; Collins, S. Requirement of vimentin filament assembly for beta3-adrenergic receptor activation of ERK MAP kinase and lipolysis. J. Biol. Chem. 2007, 282, 9244–9250. [Google Scholar] [CrossRef]
- Lieber, J.G.; Evans, R.M. Disruption of the vimentin intermediate filament system during adipose conversion of 3T3-L1 cells inhibits lipid droplet accumulation. J. Cell Sci. 1996, 109, 3047–3058. [Google Scholar]
- Thorn, H.; Stenkula, K.G.; Karlsson, M.; Ortegren, U.; Nystrom, F.H.; Gustavsson, J.; Stralfors, P. Cell surface orifices of caveolae and localization of caveolin to the necks of caveolae in adipocytes. Mol. Biol. Cell 2003, 14, 3967–3976. [Google Scholar] [CrossRef]
- Pilch, P.F.; Liu, L. Fat caves: Caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol. Metab. 2011, 22, 318–324. [Google Scholar] [CrossRef]
- Liu, L.; Brown, D.; McKee, M.; Lebrasseur, N.K.; Yang, D.; Albrecht, K.H.; Ravid, K.; Pilch, P.F. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008, 8, 310–317. [Google Scholar] [CrossRef]
- Hayashi, Y.K.; Matsuda, C.; Ogawa, M.; Goto, K.; Tominaga, K.; Mitsuhashi, S.; Park, Y.-E.; Nonaka, I.; Hino-Fukuyo, N.; Haginoya, K.; et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J. Clin. Investig. 2009, 119, 2623–2633. [Google Scholar] [CrossRef]
- Shastry, S.; Delgado, M.R.; Dirik, E.; Turkmen, M.; Agarwal, A.K.; Garg, A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am. J. Med. Genet. A 2010, 152A, 2245–2253. [Google Scholar] [CrossRef]
- Ding, S.; Lee, M.; Summer, R.; Liu, L.; Fried, S.K.; Pilch, P.F. Pleiotropic Effects of Cavin-1 Deficiency on Lipid Metabolism. J. Biol. Chem. 2014, 289, 8473–8483. [Google Scholar] [CrossRef]
- Örtegren, U.; Yin, L.; Oest, A.; Karlsson, H.; Nystrom, F.H.; Strålfors, P. Separation and characterization of caveolae subclasses in the plasma membrane of primary adipocytes; segregation of specific proteins and functions. FEBS J. 2006, 273, 3381–3392. [Google Scholar] [CrossRef]
- Aboulaich, N.; Örtegren, U.; Vener, A.V.; Strålfors, P. Association and insulin regulated translocation of hormone-sensitive lipase with PTRF. Biochem. Biophys. Res. Commun. 2006, 350, 657–661. [Google Scholar] [CrossRef]
- Zhou, S.; Guo, L.; Wang, X.; Liu, Y.; Peng, W.; Liu, Y. Acetylation of Cavin-1 Promotes Lipolysis in White Adipose Tissue. Mol. Cell. Biol. 2017, 37, 1–14. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T.; Toedter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–340. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Förster, D.; Löfgren, P. Impaired subcutaneous adipocyte lipogenesis is associated with systemic insulin resistance and increased apolipoprotein B/AI ratio in men and women. J. Intern. Med. 2007, 100, 131–139. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mairal, A.; Ghilain, C.; Mouisel, E.; Benhamed, F.; Masri, B.; Recazens, E.; Denechaud, P.; Tavernier, G.; et al. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nat. Metab. 2019, 1, 133–146. [Google Scholar] [CrossRef]
- Girousse, A.; Tavernier, G.; Valle, C.; Moro, C.; Mejhert, N.; Dinel, A.-L.; Houssier, M.; Roussel, B.; Besse-Patin, A.; Combes, M.; et al. Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biol. 2013, 11, e1001485. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Garcia-Macia, M.; Sahu, S.; Athonvarangkul, D.; Liebling, E.; Merlo, P.; Cecconi, F.; Schwartz, G.J.; Singh, R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016, 23, 113–127. [Google Scholar] [CrossRef]
- Miller, W.L.; Strauss, J.F. Molecular pathology and mechanism of action of the steroidogenic acute regulatory protein, STAR. J. Steroid Biochem. Mol. Biol. 1999, 69, 131–141. [Google Scholar] [CrossRef]
- Shen, W.J.; Patel, S.; Natu, V.; Hong, R.; Wang, J.; Azhar, S.; Kraemer, F.B. Interaction of Hormone-sensitive Lipase with Steroidogeneic Acute Regulatory Protein: Facilitation of cholesterol transfer in adrenal. J. Biol. Chem. 2003, 278, 43870–43876. [Google Scholar] [CrossRef]
- Karlsson, M.; Contreras, J.A.; Hellman, U.; Tornqvist, H.; Holm, C. cDNA Cloning, Tissue Distribution, and Identification of the Catalytic Triad of Monoglyceride Lipase. J. Biol. Chem. 1997, 272, 27218–27223. [Google Scholar] [CrossRef]
- Labar, G.; Bauvois, C.; Borel, F.; Ferrer, J.L.; Wouters, J.; Lambert, D.M. Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. ChemBioChem 2010, 11, 218–227. [Google Scholar] [CrossRef]
- Bertrand, T.; Augé, F.; Houtmann, J.; Rak, A.; Vallée, F.; Mikol, V.; Berne, P.F.; Michot, N.; Cheuret, D.; Hoornaert, C.; et al. Structural Basis for Human Monoglyceride Lipase Inhibition. J. Mol. Biol. 2010, 396, 663–673. [Google Scholar] [CrossRef]
- Karlsson, M.; Reue, K.; Xia, Y.R.; Lusis, A.J.; Langin, D.; Tornqvist, H.; Holm, C. Exon-intron organization and chromosomal localization of the mouse monoglyceride lipase gene. Gene 2001, 272, 11–18. [Google Scholar] [CrossRef]
- Tornqvist, H.; Belfrage, P. Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J. Biol. Chem. 1976, 251, 813–819. [Google Scholar]
- Savinainen, J.R.; Kansanen, E.; Pantsar, T.; Navia-Paldanius, D.; Parkkari, T.; Lehtonen, M.; Laitinen, T.; Nevalainen, T.; Poso, A.; Levonen, A.L.; et al. Robust hydrolysis of prostaglandin glycerol esters by human monoacylglycerol lipase (MAGL). Mol. Pharmacol. 2014, 86, 522–535. [Google Scholar] [CrossRef]
- Heier, C.; Taschler, U.; Radulovic, M.; Aschauer, P.; Eichmann, T.O.; Grond, S.; Wolinski, H.; Oberer, M.; Zechner, R.; Kohlwein, S.D.; et al. Monoacylglycerol lipases act as evolutionarily conserved regulators of non-oxidative ethanol metabolism. J. Biol. Chem. 2016, 291, 11865–11875. [Google Scholar] [CrossRef]
- Yoshida, K.; Kita, Y.; Tokuoka, S.M.; Hamano, F.; Yamazaki, M.; Sakimura, K.; Kano, M.; Shimizu, T. Monoacylglycerol lipase deficiency affects diet-induced obesity, fat absorption, and feeding behavior in CB1 cannabinoid receptor-deficient mice. FASEB J. 2019, 33, 2484–2497. [Google Scholar] [CrossRef]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef]
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.D.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.F.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292. [Google Scholar] [CrossRef]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef]
- Taschler, U.; Radner, F.P.W.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef]
- Chanda, P.K.; Gao, Y.; Mark, L.; Btesh, J.; Strassle, B.W.; Lu, P.; Piesla, M.J.; Zhang, M.-Y.; Bingham, B.; Uveges, A.; et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol. Pharmacol. 2010, 78, 996–1003. [Google Scholar] [CrossRef]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef]
- MacHado, J.D.; Díaz-Vera, J.; Domínguez, N.; Álvarez, C.M.; Pardo, M.R.; Borges, R. Chromogranins A and B as regulators of vesicle cargo and exocytosis. Cell. Mol. Neurobiol. 2010, 30, 1181–1187. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef]
- Rajasekaran, D.; Jariwala, N.; Mendoza, R.G.; Robertson, C.L.; Akiel, M.A.; Dozmorov, M.; Fisher, P.B.; Sarkar, D. Staphylococcal nuclease and tudor domain containing 1 (SND1 protein) promotes hepatocarcinogenesis by inhibiting monoglyceride lipase (MGLL). J. Biol. Chem. 2016, 291, 10736–10746. [Google Scholar] [CrossRef]
- Jariwal, N.; Rajasekaran, D.; Srivastava, J.; Gredler, R.; Akiel, M.A.; Robertson, C.L.; Emdad, L.; Fisher, P.B.; Sarkar, D. Role of the staphylococcal nuclease and tudor domain containing 1 in oncogenesis (Review). Int. J. Oncol. 2015, 46, 465–473. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2011, 140, 49–61. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, L.; Luo, X.; Jin, W.; He, Q.; An, J.; Lui, K.; Shi, J.; Rong, R.; Su, W.; et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene 2013, 32, 234–241. [Google Scholar] [CrossRef]
- Carbonetti, G.; Wilpshaar, T.; Kroonen, J.; Studholme, K.; Converso, C.; d’Oelsnitz, S.; Kaczocha, M. FABP5 coordinates lipid signaling that promotes prostate cancer metastasis. Sci. Rep. 2019, 9, 18944. [Google Scholar] [CrossRef]
- Schweiger, M.; Romauch, M.; Schreiber, R.; Grabner, G.F.; Hütter, S.; Kotzbeck, P.; Benedikt, P.; Eichmann, T.O.; Yamada, S.; Knittelfelder, O.; et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 2017, 8, 14859. [Google Scholar] [CrossRef]
- Ebdrup, S.; Refsgaard, H.H.F.; Fledelius, C.; Jacobsen, P. Synthesis and structure-activity relationship for a novel class of potent and selective carbamate-based inhibitors of hormone selective lipase with acute in vivo antilipolytic effects. J. Med. Chem. 2007, 50, 5449–5456. [Google Scholar] [CrossRef]
- Grabner, G.F.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride lipase as a drug target: At the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofer, P.; Taschler, U.; Schreiber, R.; Kotzbeck, P.; Schoiswohl, G. The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation. Metabolites 2020, 10, 147. https://doi.org/10.3390/metabo10040147
Hofer P, Taschler U, Schreiber R, Kotzbeck P, Schoiswohl G. The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation. Metabolites. 2020; 10(4):147. https://doi.org/10.3390/metabo10040147
Chicago/Turabian StyleHofer, Peter, Ulrike Taschler, Renate Schreiber, Petra Kotzbeck, and Gabriele Schoiswohl. 2020. "The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation" Metabolites 10, no. 4: 147. https://doi.org/10.3390/metabo10040147
APA StyleHofer, P., Taschler, U., Schreiber, R., Kotzbeck, P., & Schoiswohl, G. (2020). The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation. Metabolites, 10(4), 147. https://doi.org/10.3390/metabo10040147