Identification of Metabolic Alterations in Breast Cancer Using Mass Spectrometry-Based Metabolomic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. GCTOF MS Analysis
2.2.1. Data Acquisition
2.2.2. Data Processing
2.3. HILIC-ESI-QTOF-MS/MS Analysis
2.3.1. Data Acquisition
2.3.2. Data Processing
2.4. Bioinformatics Analysis for Identification of Metabolic Marker Candidates
2.5. Statistical Analysis
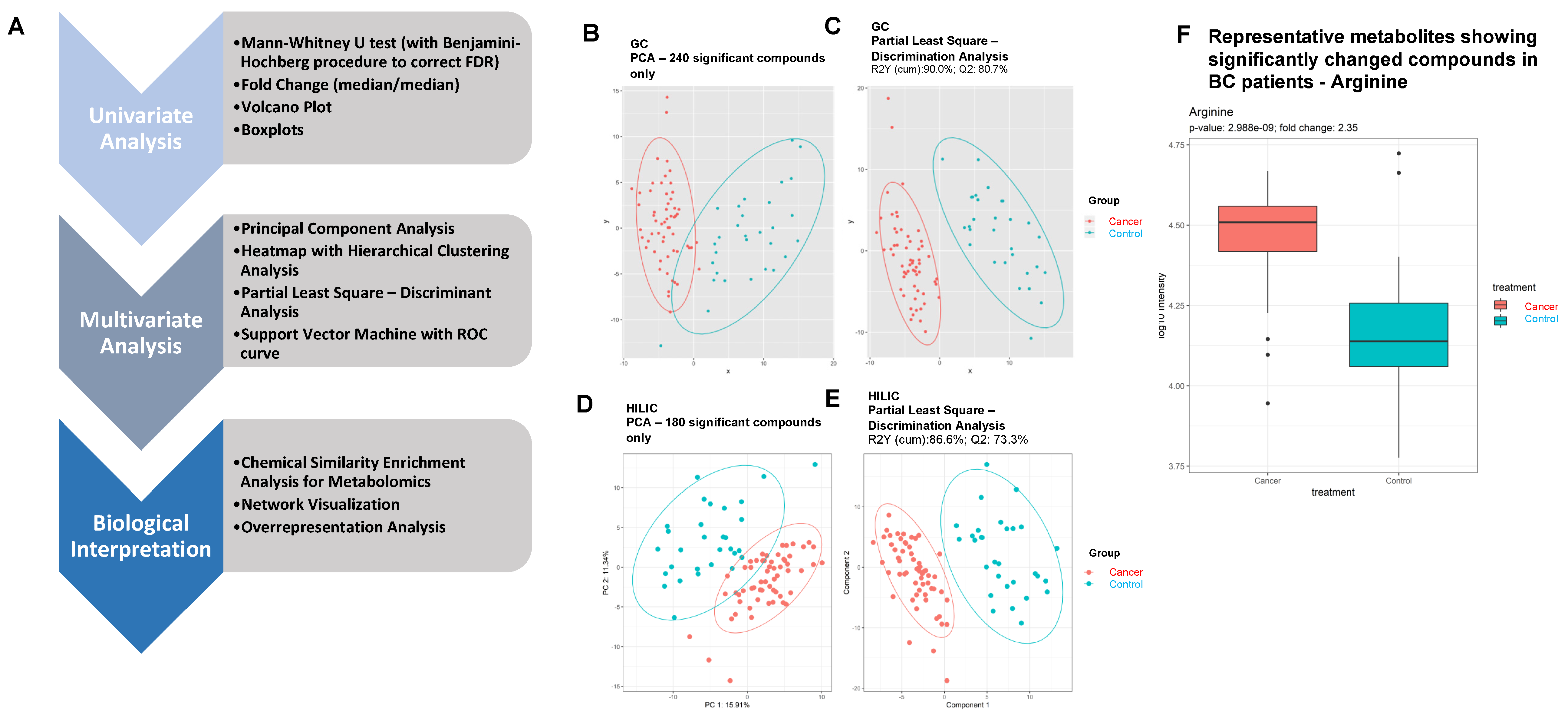
2.5.1. Univariate Analysis
2.5.2. Multivariate Analysis
2.6. Biological Interpretation
3. Results
3.1. Characteristics of the Study Subjects
3.2. Primary Metabolomics Analyses Were Performed via ALEX-CIS-GC-TOF-MS and HILIC-ESI-QTOF-MS/MS
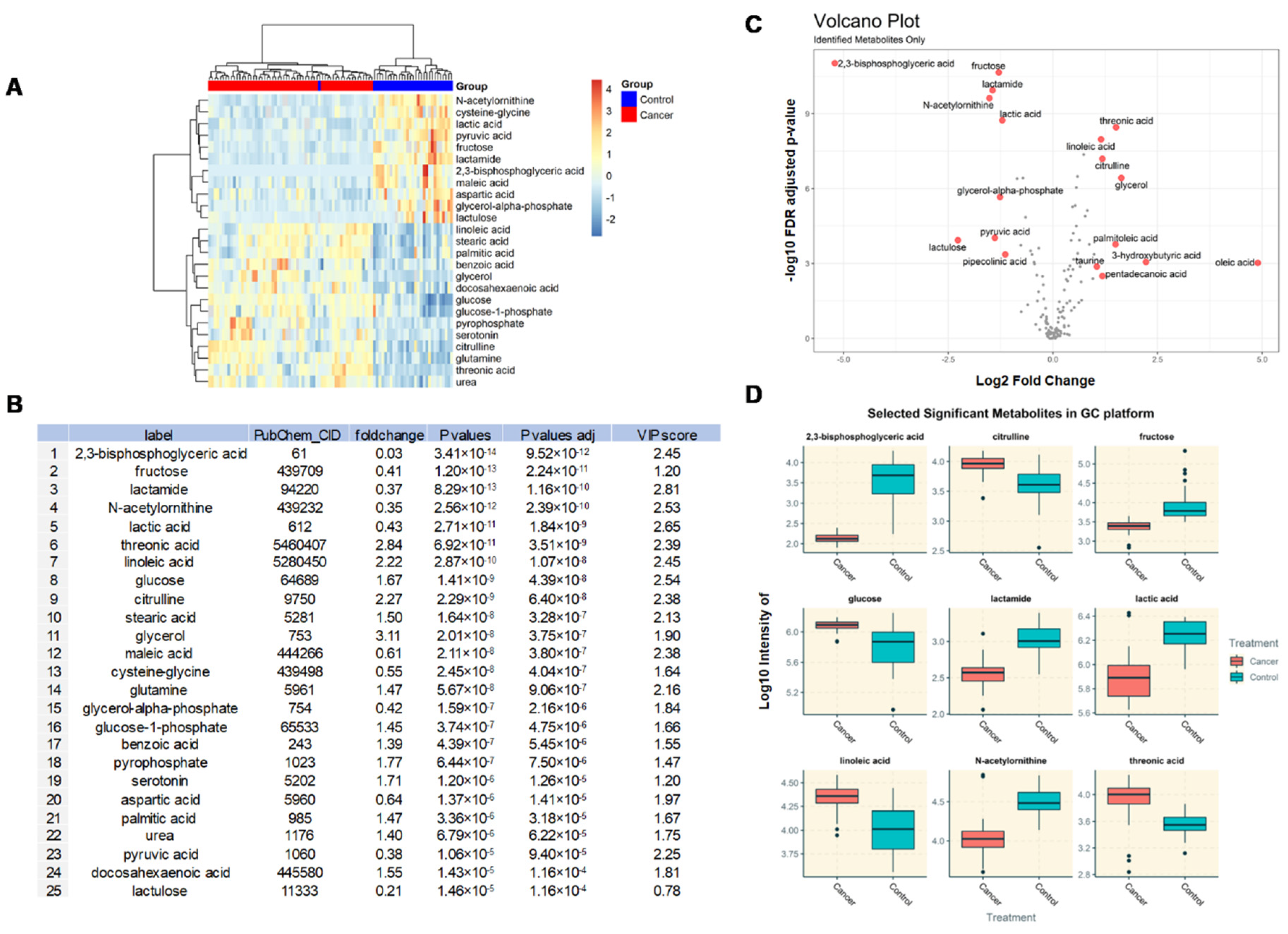
3.3. Identification of DEMs in BC Patients
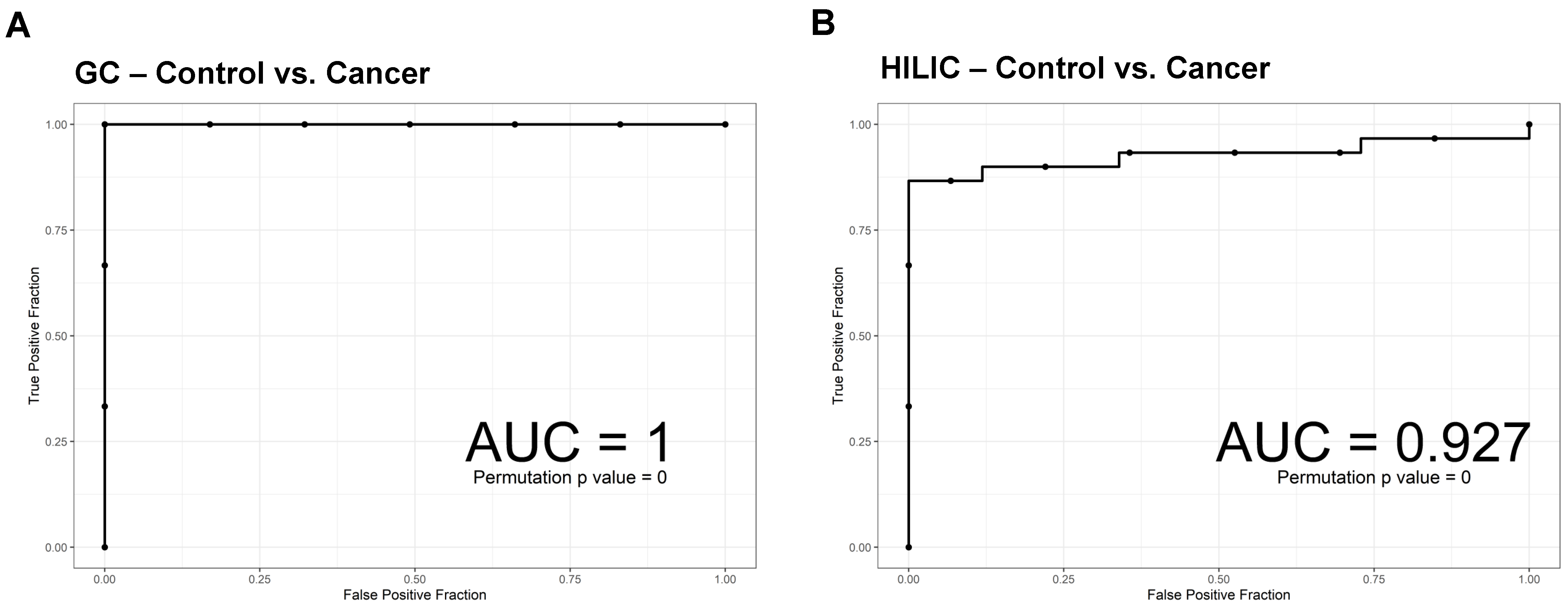
3.4. ROC Curves of SVM.
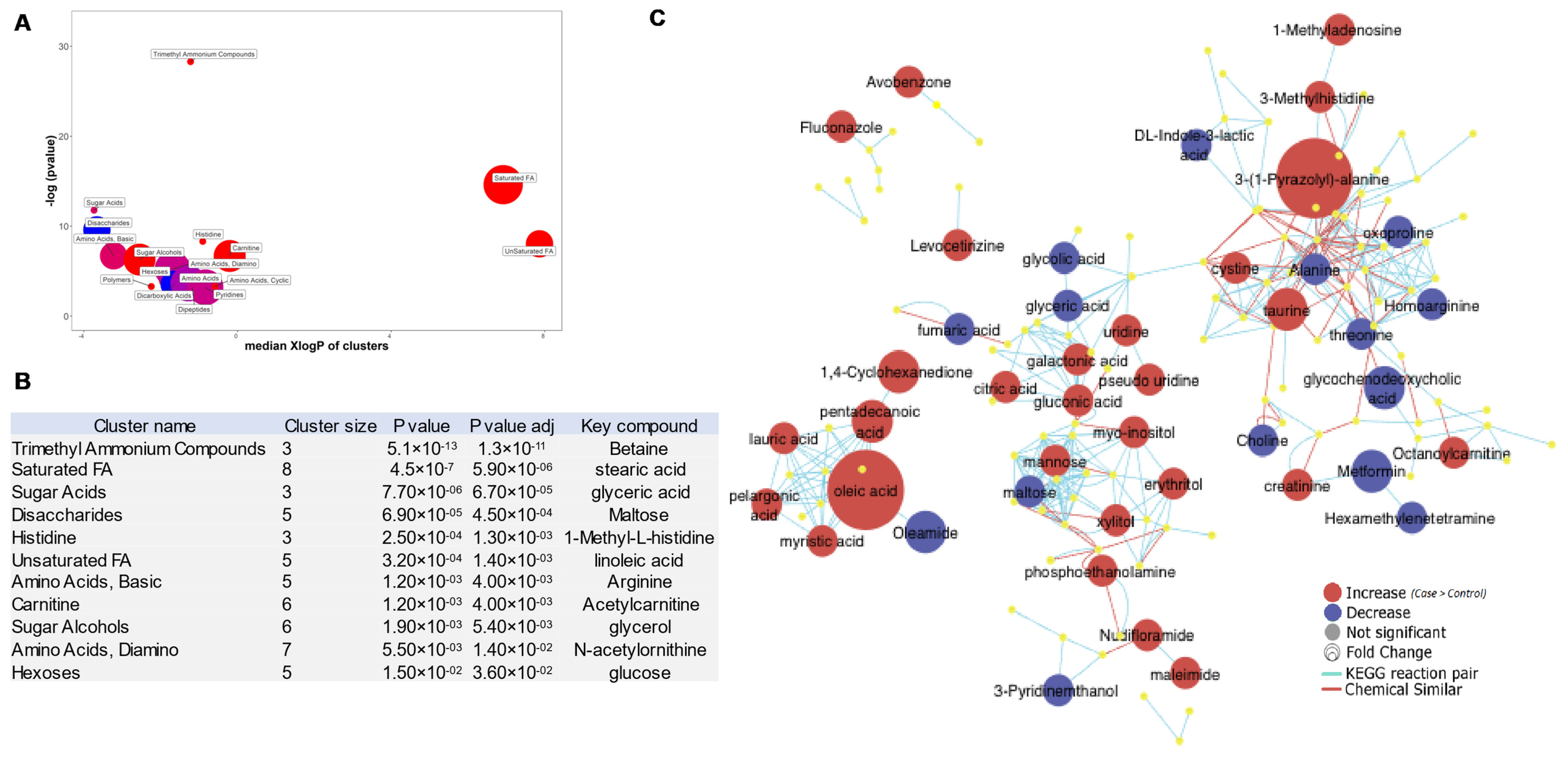
3.5. ChemRICH Plots of BC-Associated Metabolites
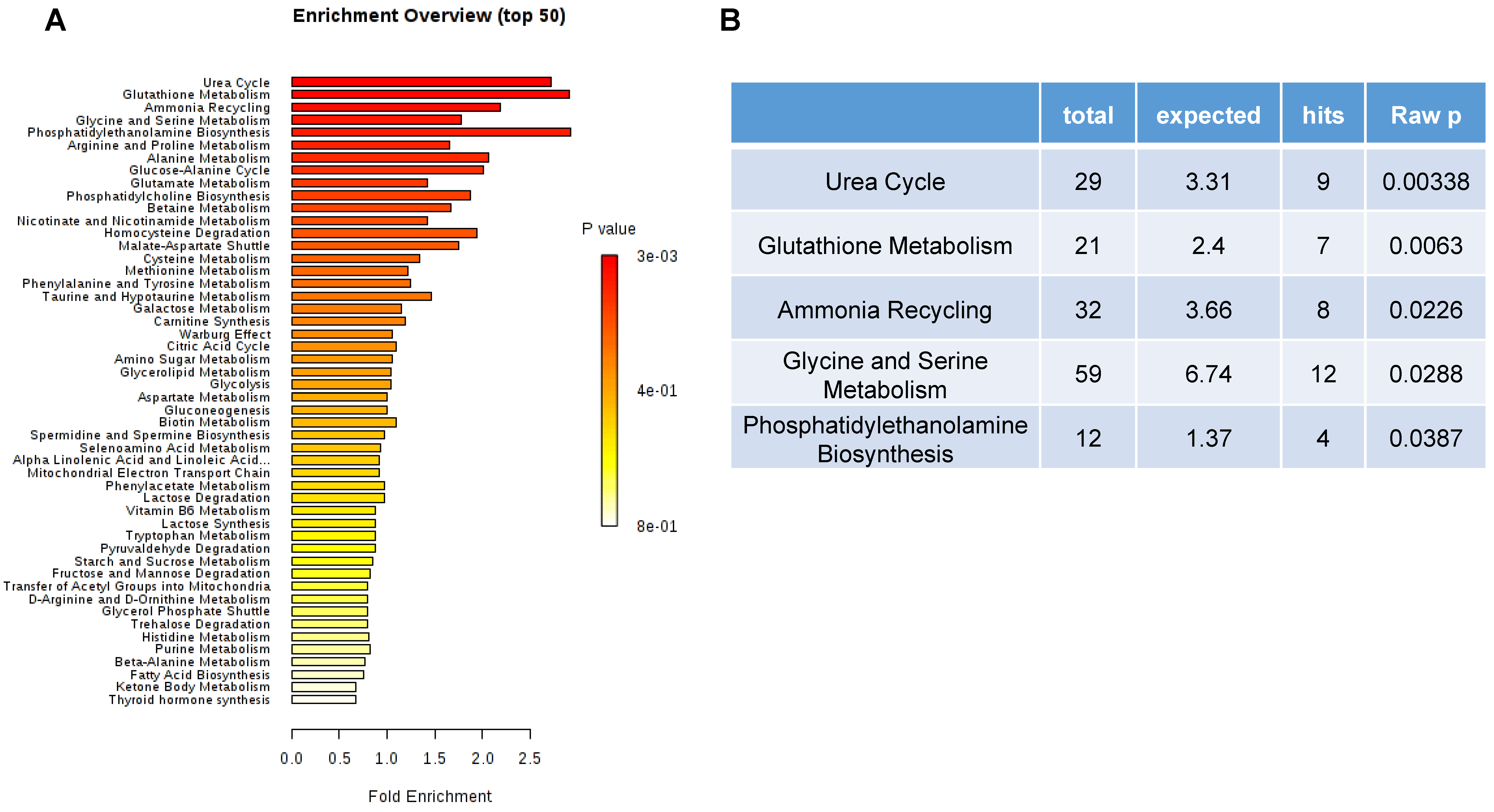
3.6. Top 50 Metabolic Pathway-Associated DEMs Sets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, S.H.; Saadatpour, Z.; Salmaninejad, A.; Momeni, F.; Mokhtari, M.; Nahand, J.S.; Rahmati, M.; Mirzaei, H.; Kianmehr, M. Breast cancer diagnosis: Imaging techniques and biochemical markers. J. Cell Physiol. 2018, 233, 5200–5213. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Early Diagnosis of Breast Cancer. Sensors 2017, 17, 1572. [Google Scholar] [CrossRef] [PubMed]
- Donepudi, M.S.; Kondapalli, K.; Amos, S.J.; Venkanteshan, P. Breast cancer statistics and markers. J. Cancer Res. Ther. 2014. [Google Scholar] [CrossRef]
- Duffy, M.J. Serum Tumor Markers in Breast Cancer: Are They of Clinical Value? Clin. Chem. 2006, 52, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [Green Version]
- Hart, C.D.; Tenori, L.; Luchinat, C.; Di Leo, A. Metabolomics in Breast Cancer: Current Status and Perspectives. Adv. Exp. Med. Biol. 2016, 882, 217–234. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Network, C.G.A.; Network, T.C.G.A.; Koboldt, D.C.; Fulton, R.S.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Bathen, T.F.; Sitter, B.; Tessem, M.-B.; Gribbestad, I.S.; Sjøbakk, T.E. Magnetic Resonance Metabolomics of Intact Tissue: A Biotechnological Tool in Cancer Diagnostics and Treatment Evaluation. Cancer Res. 2010, 70, 6692–6696. [Google Scholar] [CrossRef] [Green Version]
- Coleman, W.B. Next-Generation Breast Cancer Omics. Am. J. Pathol. 2017, 187, 2130–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, Y.J.; Kim, E.-S.; Koo, J.S. Amino Acid Transporters and Glutamine Metabolism in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to “Glutamine Addiction”: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, S.; Bayerlová, M.; Vetter, M.; Wachter, A.; Mitra, D.; Hanf, V.; Lantzsch, T.; Uleer, C.; Peschel, S.; John, J.; et al. Proteomic profiling of breast cancer metabolism identifies SHMT2 and ASCT2 as prognostic factors. Breast Cancer Res. 2017, 19, 112. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, S.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Babu, E.; Periyasamy-Thandavan, S.; Gurav, A.; Gnanaprakasam, J.P.; Singh, N.; Schoenlein, P.V.; et al. SLC6A14 (ATB0,+) Protein, a Highly Concentrative and Broad Specific Amino Acid Transporter, Is a Novel and Effective Drug Target for Treatment of Estrogen Receptor-positive Breast Cancer. J. Boil. Chem. 2011, 286, 31830–318384. [Google Scholar] [CrossRef] [Green Version]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2011, 103, 382–389. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Schuster, D.M. Amino Acid Metabolism as a Target for Breast Cancer Imaging. PET Clin. 2018, 13, 437–444. [Google Scholar] [CrossRef]
- Leskinen-Kallio, S.; Någren, K.; Lehikoinen, P.; Ruotsalainen, U.; Joensuu, H. Uptake of 11C-methionine in breast cancer studied by PET. An association with the size of S-phase fraction. Br. J. Cancer 1991, 64, 1121–1124. [Google Scholar] [CrossRef] [Green Version]
- Tade, F.I.; Cohen, M.; Styblo, T.M.; Odewole, O.A.; Holbrook, A.I.; Newell, M.S.; Savir-Baruch, B.; Li, X.B.; Goodman, M.M.; A Nye, J.; et al. Anti-3-[18F] FACBC (Fluciclovine) PET-CT of Breast Cancer An Exploratory Study. J. Nucl. Med. 2016, 57, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Haukaas, T.H.; Euceda, L.R.; Giskeødegård, G.F.; Bathen, T.F. Metabolic Portraits of Breast Cancer by HR MAS MR Spectroscopy of Intact Tissue Samples. Metabolites 2017, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Tolstikov, V.; Fiehn, O.; Weiss, R. A comprehensive urinary metabolomic approach for identifying kidney cancer. Anal. Biochem. 2007, 363, 185–195. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [Green Version]
- Szymańska, E.; Saccenti, E.; Smilde, A.K.; Westerhuis, J.A. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 2012, 8 (Suppl. 1), 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.D.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiehn, O.; Wohlgemuth, G.; Scholz, M.; Kind, T.; Lee, Y.; Lu, Y.; Moon, S.; Nikolau, B. Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 2008, 53, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Barupal, D.K.; Fiehn, O. Chemical Similarity Enrichment Analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 2017, 7, 14567. [Google Scholar] [CrossRef]
- Barupal, D.K.; Haldiya, P.K.; Wohlgemuth, G.; Kind, T.; Kothari, S.L.; Pinkerton, K.E.; Fiehn, O. MetaMapp: Mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinform. 2012, 13, 99. [Google Scholar] [CrossRef] [Green Version]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2015, 35, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis Activates Rag-mTORC1 Signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappelletti, V.; Iorio, E.; Miodini, P.; Silvestri, M.; Dugo, M.; Daidone, M.G. Metabolic Footprints and Molecular Subtypes in Breast Cancer. Dis. Markers 2017, 2017, 7687851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budczies, J.; Brockmöller, S.; Müller, B.M.; Barupal, D.K.; Richter-Ehrenstein, C.; Kleine-Tebbe, A.; Griffin, J.L.; Orešič, M.; Dietel, M.; Denkert, C.; et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: Alterations in glutamine and beta-alanine metabolism. J. Proteom. 2013, 94, 279–288. [Google Scholar] [CrossRef]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. JBIC J. Boil. Inorg. Chem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- A Barzał, J.; Szczylik, C.; Rzepecki, P.; Jaworska, M.; Anuszewska, E. Plasma citrulline level as a biomarker for cancer therapy-induced small bowel mucosal damage. Acta Biochim. Pol. 2014, 61. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, N.; Das, G. Metabolic Reprogramming in Breast Cancer and Its Therapeutic Implications. Cells 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, M.-Y.; Park, S.W.; Moon, J.-S.; Koh, Y.-K.; Ahn, Y.H.; Park, S.; Kim, K.-S. Up-regulation of Acetyl-CoA Carboxylase α and Fatty Acid Synthase by Human Epidermal Growth Factor Receptor 2 at the Translational Level in Breast Cancer Cells. J. Boil. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Geck, R.C.; Foley, J.R.; Stewart, T.M.; Asara, J.M.; Casero, R.A., Jr.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Boil. Chem. 2020. [Google Scholar] [CrossRef] [Green Version]
- Geck, R.C.; Toker, A. Nonessential amino acid metabolism in breast cancer. Adv. Boil. Regul. 2016, 62, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tito, N.; Guzman, A.S.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes migration on MDA-MB-231 breast cancer cells through an arachidonic acid-dependent pathway. Int. J. Biochem. Cell Boil. 2010, 42, 306–317. [Google Scholar] [CrossRef]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar] [PubMed]
- Marcial-Medina, C.; Ordoñez-Moreno, A.; Gonzalez-Reyes, C.; Cortes-Reynosa, P.; Salazar, E.P. Oleic acid induces migration through a FFAR1/4, EGFR and AKT-dependent pathway in breast cancer cells. Endocr. Connect. 2019, 8, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, S.; Riedel, S.; Gelderblom, W.C.A. Dietary PUFA and cancer. Proc. Nutr. Soc. 2014, 73, 361–367. [Google Scholar] [CrossRef]
- Serna-Marquez, N.; Diaz-Aragon, R.; Reyes-Uribe, E.; Cortes-Reynosa, P.; Salazar, E.P. Linoleic acid induces migration and invasion through FFAR4- and PI3K-/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Med. Oncol. 2017, 34, 45. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, S.; Shahid, M.; Jin, P.; Asher, A.; Kim, J. Identification of Metabolic Alterations in Breast Cancer Using Mass Spectrometry-Based Metabolomic Analysis. Metabolites 2020, 10, 170. https://doi.org/10.3390/metabo10040170
Fan S, Shahid M, Jin P, Asher A, Kim J. Identification of Metabolic Alterations in Breast Cancer Using Mass Spectrometry-Based Metabolomic Analysis. Metabolites. 2020; 10(4):170. https://doi.org/10.3390/metabo10040170
Chicago/Turabian StyleFan, Sili, Muhammad Shahid, Peng Jin, Arash Asher, and Jayoung Kim. 2020. "Identification of Metabolic Alterations in Breast Cancer Using Mass Spectrometry-Based Metabolomic Analysis" Metabolites 10, no. 4: 170. https://doi.org/10.3390/metabo10040170