The Key Role of Purine Metabolism in the Folate-Dependent Phenotype of Autism Spectrum Disorders: An In Silico Analysis

 ,
,
Abstract
:1. Introduction
- The production of methionine from homocysteine, followed by its conversion to S-adenosylmethionine (SAM), which primarily provides methyl groups for DNA, RNA, chromatin, protein, and phospholipid methylation, in order to maintain the physiological regulation of gene expression during brain development and maturation. Altered DNA methylation has been connected to ASD [23], as well as mutations in subunits of DNA methyltransferase (DNMT1, DNMT3A, DNMT3B, and DNMT3) [24,25,26];
- Homocysteine degradation to cystathionine and consecutive glutathione synthesis (transsulfuration pathway) produces protective factors against reactive oxygen species (ROS). A reduction in the methionine regeneration cycle also results in a reduction in antioxidant synthesis activity [27]. In the case of folate depletion, methionine can be regenerated by an alternative pathway that converts choline to betaine and then to dimethylglycine via betaine homocysteine methyltransferase (BHMT) [28]. Lowered levels of betaine have been found in ASD [29];
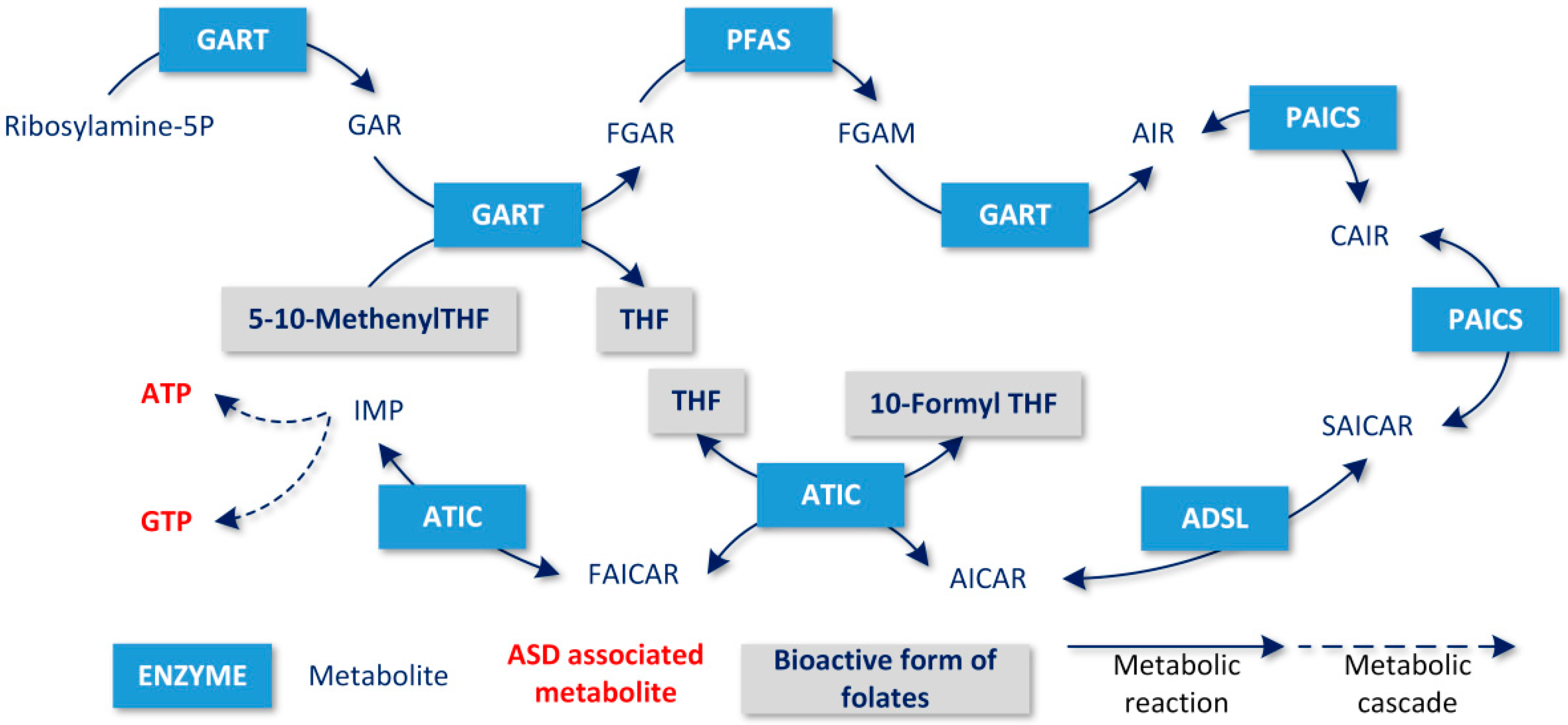
- The production of tetrahydrobiopterin (BH4) via the synthesis of purines is a substrate for the precursors of catecholamines. Hyperserotonemia has been associated with the induction of ASD in animal models [30]; conversely, melatonin levels decrease, and its compensation has been clinically studied in ASD [31]. BH4 also enables the synthesis of l-3,4-dihydroxyphenylalanine (L-DOPA) and a recent study showed abnormal levels of dopamine in patients with ASD [32]. The synthesis of purines utilizes adenosine, which is a byproduct of the methionine regeneration cycle [33];
- BH4 is also necessary for the synthesis of nitric oxide (NO). Under healthy conditions, BH4 is regenerated, but if the concentration of BH4 is lowered, peroxynitrite (ONOOH) is produced. Its accumulation leads to the hyperexcitation of NMDA receptors, and together with the accumulation of homocysteine, induces the apoptosis of neurons [15]. Oxidized pterins inhibit NO synthase and further lower the synthesis of NO [33];
- Synthesis of pyrimidine as a substrate for DNA synthesis and replication.
2. Results
2.1. Blocked Metabolites Associated with Gene Knockouts
2.2. Systemic Folate Deficiency from the Viewpoint of Blocked Metabolites
2.3. Cerebral Folate Deficiency from the Viewpoint of Blocked Metabolites
2.4. Implications for the Pathophysiology of ASD
3. Discussion
4. Materials and Methods
4.1. Flux Balance Analysis Terminology
4.2. Blocked Reactions and Metabolites
4.3. Measurements of Blocked Metabolite Set Overlap
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A. Code Availability
Appendix B. Datasets Used
- -
- Recon 2.2: The model is freely available from the Biomodels database, under the identifier MODEL1603150001 [51]. The model is available at http://identifiers.org/biomodels.db/MODEL1603150001.
- -
- An automatically generated metabolic model of the cerebral cortex neuron using the CORDA method [65]. The model is available at https://qutublab.org/apps-code-tools/#Metabolic.
References
- Chaaya, M.; Saab, D.; Maalouf, F.T.; Boustany, R.M. Prevalence of Autism Spectrum Disorder in Nurseries in Lebanon: A Cross Sectional Study. J. Autism Dev. Disord. 2016, 46, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Crespo, L.; Prats-Uribe, A.; Tobias, A.; Duran-Tauleria, E.; Coronado, R.; Hervás, A.; Guxens, M. Temporal and Geographical Variability of Prevalence and Incidence of Autism Spectrum Disorder Diagnoses in Children in Catalonia, Spain. Autism Res. 2019, 12, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baio, J. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014, 63, 1–21. [Google Scholar]
- Diallo, F.B.; Fombonne, É.; Kisely, S.; Rochette, L.; Vasiliadis, H.M.; Vanasse, A.; Noiseux, M.; Pelletier, É.; Renaud, J.; St-Laurent, D.; et al. Prevalence and Correlates of Autism Spectrum Disorders in Quebec: Prévalence et corrélats des troubles du spectre de l’autisme au Québec. Can. J. Psychiatry 2018, 63, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Barrett, B. Substantial lifelong cost of autism spectrum disorder. J. Pediatr. 2014, 165, 1068–1069. [Google Scholar] [CrossRef]
- Heath, A.K.; Ganz, J.B.; Parker, R.; Burke, M.; Ninci, J. A Meta-analytic Review of Functional Communication Training Across Mode of Communication, Age, and Disability. Rev. J. Autism Dev. Disord. 2015, 2, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Richman, D.M.; Barnard-Brak, L.; Grubb, L.; Bosch, A.; Abby, L. Meta-analysis of noncontingent reinforcement effects on problem behavior. J. Appl. Behav. Anal. 2015, 48, 131–152. [Google Scholar] [CrossRef]
- Kurtz, P.F.; Chin, M.D.; Huete, J.M.; Tarbox, R.S.F.; O’Connor, J.T.; Paclawskyj, T.R.; Rush, K.S. Functional analysus and treatment of self-injurious behavior in young children: A summary of 30 cases. J. Appl. Behav. Anal. 2003, 36, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.L.M.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Sequeira, J.M.; Blau, N.; Quadros, E.V. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev. Med. Child. Neurol. 2008, 50, 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modabbernia, A.; Velthorst, E.; Reichenberg, A. Environmental risk factors for autism: An evidence-based review of systematic reviews and meta-analyses. Mol. Autism 2017, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef]
- Wang, M.; Li, K.; Zhao, D.; Li, L. The association between maternal use of folic acid supplements during pregnancy and risk of autism spectrum disorders in children: A meta-analysis. Mol. Autism 2017, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Surén, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, V.T.; Sequeira, J.M.; Quadros, E.V. The basis for folinic acid treatment in neuro-psychiatric disorders. Biochimie 2016, 126, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Shen, Y.; Wu, J. Association between MTHFR gene polymorphisms and the risk of autism spectrum disorders: A meta-analysis. Autism Res. 2013, 6, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Cario, H.; Smith, D.E.C.; Blom, H.; Blau, N.; Bode, H.; Holzmann, K.; Pannicke, U.; Hopfner, K.P.; Rump, E.M.; Ayric, Z.; et al. Dihydrofolate reductase deficiency due to a homozygous DHFR mutation causes megaloblastic anemia and cerebral folate deficiency leading to severe neurologic disease. Am. J. Hum. Genet. 2011, 88, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Farsi, Y.M.; Waly, M.I.; Deth, R.C.; Al-Sharbati, M.M.; Al-Shafaee, M.; Al-Farsi, O.; Al-Khaduri, M.M.; Gupta, I.; Ali, A.; Al-Khalili, M.; et al. Low folate and vitamin B12 nourishment is common in Omani children with newly diagnosed autism. Nutrition 2013, 29, 537–541. [Google Scholar] [CrossRef]
- Zhao, R.; Visentin, M.; Suadicani, S.O.; Goldman, I.D. Inhibition of the proton-coupled folate transporter (PCFT-SLC46A1) by bicarbonate and other anions. Mol. Pharmacol. 2013, 84, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Ramaekers, V.T.T.; Segers, K.; Sequeira, J.M.M.; Koenig, M.; Van Maldergem, L.; Bours, V.; Kornak, U.; Quadros, E.V.V. Genetic assessment and folate receptor autoantibodies in infantile-onset cerebral folate deficiency (CFD) syndrome. Mol. Genet. Metab. 2018, 124, 87–93. [Google Scholar] [CrossRef]
- Frye, R.E.; Delhey, L.; Slattery, J.; Tippett, M.; Wynne, R.; Rose, S.; Kahler, S.G.; Bennuri, S.C.; Melnyk, S.; Sequeira, J.M.; et al. Blocking and Binding Folate Receptor Alpha Autoantibodies Identify Novel Autism Spectrum Disorder Subgroups. Front. Neurosci. 2016, 10, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol. Psychiatry 2013, 18, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Hogart, A.R.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, e1–e11. [Google Scholar] [CrossRef] [Green Version]
- Alex, A.M.; Saradalekshmi, K.R.; Shilen, N.; Suresh, P.A.; Banerjee, M. Genetic association of DNMT variants can play a critical role in defining the methylation patterns in autism. IUBMB Life 2019, 71, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [Green Version]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Schaevitz, L.R.; Berger-Sweeney, J.E. Gene-environment interactions and epigenetic pathways in autism: The importance of one-carbon metabolism. ILAR J. 2012, 53, 322–340. [Google Scholar] [CrossRef] [Green Version]
- Hamlin, J.C.; Pauly, M.; Melnyk, S.; Pavliv, O.; Starrett, W.; Crook, T.A.; James, S.J. Dietary intake and plasma levels of choline and betaine in children with autism spectrum disorders. Autism Res. Treat. 2013, 2013, 578429. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Sato, A.; Kasai, S.; Hagino, Y.; Kotajima-Murakami, H.; Kashii, H.; Takamatsu, Y.; Nishito, Y.; Inagaki, M.; Mizuguchi, M.; et al. Brain hyperserotonemia causes autism-relevant social deficits in mice. Mol. Autism 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Melatonin in autism spectrum disorders: A systematic review and meta-analysis. Dev. Med. Child. Neurol. 2011, 53, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Esmaiel, N.N.; Ashaat, E.A.; Mosaad, R.; Fayez, A.; Ibrahim, M.; Abdallah, Z.Y.; Issa, M.Y.; Salem, S.; Ramadan, A.; El Wakeel, M.A.; et al. The potential impact of COMT gene variants on dopamine regulation and phenotypic traits of ASD patients. Behav. Brain Res. 2020, 378, 112272. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; DeLatorre, R.; Taylor, H.B.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Metabolic effects of sapropterin treatment in autism spectrum disorder: A preliminary study. Transl. Psychiatry 2013, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K. Antipurinergic therapy for autism-An in-depth review. Mitochondrion 2018, 43, 1–15. [Google Scholar] [CrossRef]
- Naviaux, J.C.; Wang, L.; Li, K.; Bright, A.; Alaynick, W.A.; Williams, K.R.; Powell, S.B.; Naviaux, R.K. Antipurinergic therapy corrects the autism-like features in the Fragile X (Fmr1 knockout) mouse model. Mol. Autism 2015, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Naviaux, J.C.; Schuchbauer, M.A.; Li, K.; Wang, L.; Risbrough, V.B.; Powell, S.B.; Naviaux, R.K. Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy. Transl. Psychiatry 2014, 4, e400. [Google Scholar] [CrossRef]
- Howsmon, D.P.; Kruger, U.; Melnyk, S.; James, S.J.; Hahn, J. Classification and adaptive behavior prediction of children with autism spectrum disorder based upon multivariate data analysis of markers of oxidative stress and DNA methylation. PLoS Comput. Biol. 2017, 13, e1005385. [Google Scholar] [CrossRef]
- Li, G.; Lee, O.; Rabitz, H. High efficiency classification of children with autism spectrum disorder. PLoS ONE 2018, 13, e0192867. [Google Scholar] [CrossRef]
- Hollowood, K.; Melnyk, S.; Pavliv, O.; Evans, T.; Sides, A.; Schmidt, R.J.; Hertz-Picciotto, I.; Elms, W.; Guerrero, E.; Kruger, U.; et al. Maternal metabolic profile predicts high or low risk of an autism pregnancy outcome. Res. Autism Spectr. Disord. 2018, 56, 72–82. [Google Scholar] [CrossRef]
- Vargason, T.; Kruger, U.; Roth, E.; Delhey, L.M.; Tippett, M.; Rose, S.; Bennuri, S.C.; Slattery, J.C.; Melnyk, S.; James, S.J.; et al. Comparison of Three Clinical Trial Treatments for Autism Spectrum Disorder Through Multivariate Analysis of Changes in Metabolic Profiles and Adaptive Behavior. Front. Cell. Neurosci. 2018, 12, 503. [Google Scholar] [CrossRef] [Green Version]
- Vargason, T.; Howsmon, D.P.; Melnyk, S.; James, S.J.; Hahn, J. Mathematical modeling of the methionine cycle and transsulfuration pathway in individuals with autism spectrum disorder. J. Theor. Biol. 2017, 416, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Covert, M.; Palsson, B. Metabolic modelling of microbes: The flux-balance approach. Environ. Microbiol. 2002, 4, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Gianchandani, E.P.; Papin, J.A. Flux balance analysis in the era of metabolomics. Brief. Bioinform. 2006, 7, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Drozdov-Tikhomirov, L.N.; Scurida, G.I.; Davidov, A.V.; Alexandrov, A.A.; Zvyagilskaya, R.A. Mathematical modeling of living cell metabolism using the method of steady-state stoichiometric flux balance. J. Bioinform. Comput. Biol. 2006, 4, 865–885. [Google Scholar] [CrossRef]
- Sahoo, S.; Franzson, L.; Jonsson, J.J.; Thiele, I. A compendium of inborn errors of metabolism mapped onto the human metabolic network. Mol. Biosyst. 2012, 8, 2545–2558. [Google Scholar] [CrossRef] [Green Version]
- Karlstädt, A.; Fliegner, D.; Kararigas, G.; Ruderisch, H.S.; Regitz-Zagrosek, V.; Holzhütter, H.G. CardioNet: A human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst. Biol. 2012, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinform. 2010, 11, 489. [Google Scholar] [CrossRef] [Green Version]
- Massaiu, I.; Pasotti, L.; Sonnenschein, N.; Rama, E.; Cavaletti, M.; Magni, P.; Calvio, C.; Herrgård, M.J. Integration of enzymatic data in Bacillus subtilis genome-scale metabolic model improves phenotype predictions and enables in silico design of poly-γ-glutamic acid production strains. Microb. Cell Fact. 2019, 18, 3. [Google Scholar] [CrossRef]
- Krsička, D.; Geryk, J.; Vlčková, M.; Havlovicová, M.; Macek, M.; Pourová, R. Identification of likely associations between cerebral folate deficiency and complex genetic- and metabolic pathogenesis of autism spectrum disorders by utilization of a pilot interaction modeling approach. Autism Res. 2017, 10, 1424–1435. [Google Scholar] [CrossRef]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M.; et al. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 109. [Google Scholar] [CrossRef]
- Smart, A.G.; Amaral, L.A.N.; Ottino, J.M. Cascading failure and robustness in metabolic networks. Proc. Natl. Acad. Sci. USA 2008, 105, 13223–13228. [Google Scholar] [CrossRef] [Green Version]
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant ADSL enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455. [Google Scholar] [CrossRef]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.F. AICA-ribosiduria: A novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef] [Green Version]
- Blau, N.; Ichinose, H.; Nagatsu, T.; Heizmann, C.W.; Zacchello, F.; Burlina, A.B. A missense mutation in a patient with guanosine triphosphate cyclohydrolase I deficiency missed in the newborn screening program. J. Pediatr. 1995, 126, 401–405. [Google Scholar] [CrossRef]
- Chien, Y.H.; Chiang, S.C.; Huang, A.; Lin, J.M.; Chiu, Y.N.; Chou, S.P.; Chu, S.Y.; Wang, T.R.; Hwu, W.L. Treatment and outcome of Taiwanese patients with 6-pyruvoyltetrahydropterin synthase gene mutations. J. Inherit. Metab. Dis. 2001, 24, 815–823. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Willemsen, M.A.A.P.; Wevers, R.A.; Lagerwerf, A.J.; Abeling, N.G.G.M.; Blau, N.; Thöny, B.; Vargiami, E.; Zafeiriou, D.I. Two Greek siblings with sepiapterin reductase deficiency. Mol. Genet. Metab. 2008, 94, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Keil, K.P.; Lein, P.J. DNA methylation: A mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenetics 2016, 2, dvv012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnyk, S.; Fuchs, G.J.; Schulz, E.; Lopez, M.; Kahler, S.G.; Fussell, J.J.; Bellando, J.; Pavliv, O.; Rose, S.; Seidel, L.; et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J. Autism Dev. Disord. 2012, 42, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymańska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.K.S.; Lee, D.Y. Flux-sum analysis: A metabolite-centric approach for understanding the metabolic network. BMC Syst. Biol. 2009, 3, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, O.D.K.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krsička, D.; Vlčková, M.; Havlovicová, M. The Significance of Cerebral Folate Deficiency for the Development and Treatment of Autism Spectrum Disorders. Int. J. Biomed. Healthc. 2014, 2. [Google Scholar]
- Schultz, A.; Qutub, A.A. Reconstruction of Tissue-Specific Metabolic Networks Using CORDA. PLoS Comput. Biol. 2016, 12, e1004808. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene Name | Supercell (All Cells) | Cerebral Cortex Neuron Only | ||
|---|---|---|---|---|
| GART | 0.9762 | 0.9765 | 0.9028 | 0.8862 |
| PFAS | 0.9759 | 0.9745 | 0.9026 | 0.8861 |
| PPAT | 0.9647 | 0.9650 | 0.9016 | 0.8851 |
| PAICS | 0.9518 | 0.9509 | 0.9000 | 0.8835 |
| ADSL | 0.9277 | 0.9268 | 0.8961 | 0.8796 |
| ATIC | 0.9036 | 0.9027 | 0.8987 | 0.8822 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geryk, J.; Krsička, D.; Vlčková, M.; Havlovicová, M.; Macek, M., Jr.; Kremlíková Pourová, R. The Key Role of Purine Metabolism in the Folate-Dependent Phenotype of Autism Spectrum Disorders: An In Silico Analysis. Metabolites 2020, 10, 184. https://doi.org/10.3390/metabo10050184
Geryk J, Krsička D, Vlčková M, Havlovicová M, Macek M Jr., Kremlíková Pourová R. The Key Role of Purine Metabolism in the Folate-Dependent Phenotype of Autism Spectrum Disorders: An In Silico Analysis. Metabolites. 2020; 10(5):184. https://doi.org/10.3390/metabo10050184
Chicago/Turabian StyleGeryk, Jan, Daniel Krsička, Markéta Vlčková, Markéta Havlovicová, Milan Macek, Jr., and Radka Kremlíková Pourová. 2020. "The Key Role of Purine Metabolism in the Folate-Dependent Phenotype of Autism Spectrum Disorders: An In Silico Analysis" Metabolites 10, no. 5: 184. https://doi.org/10.3390/metabo10050184
APA StyleGeryk, J., Krsička, D., Vlčková, M., Havlovicová, M., Macek, M., Jr., & Kremlíková Pourová, R. (2020). The Key Role of Purine Metabolism in the Folate-Dependent Phenotype of Autism Spectrum Disorders: An In Silico Analysis. Metabolites, 10(5), 184. https://doi.org/10.3390/metabo10050184