A High-Throughput Method for the Comprehensive Analysis of Terpenes and Terpenoids in Medicinal Cannabis Biomass


Abstract
:
1. Introduction
2. Results
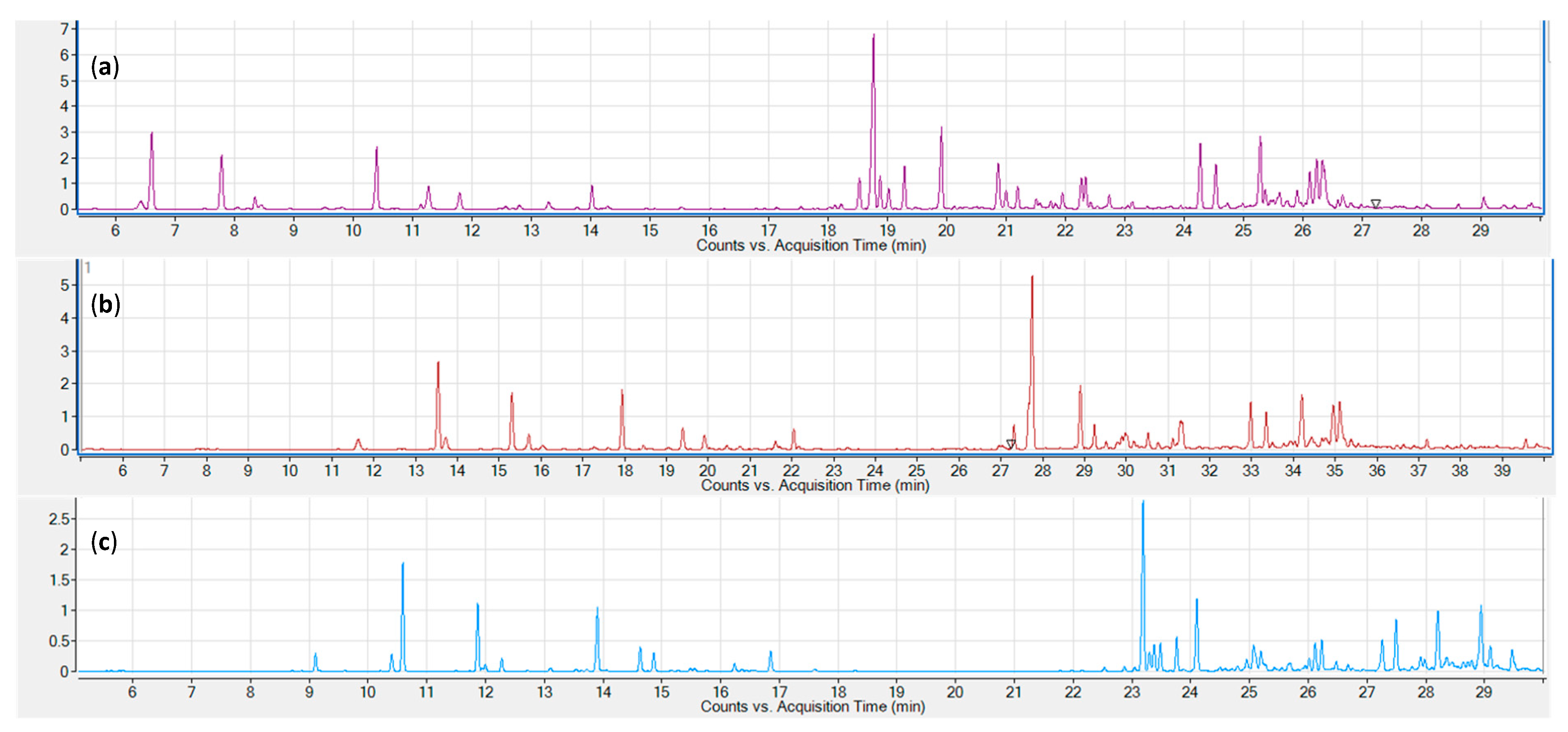
2.1. Sampling Techniques
2.2. Columns
2.3. Solvent Optimisation
2.4. Compound Identification and Resolution
2.5. Linearity
2.6. Detection and Quantitation Limits
2.7. Accuracy
2.8. Precision and Intermediate Precision
3. Discussion
- Comprehensive—detecting the highest possible number of individual compounds.
- Quantitative—reliably providing absolute quantitation for as many compounds as possible.
- High-throughput—processing the maximum number of samples possible in as short a time as possible, from as little biomass as possible.
4. Materials and Methods
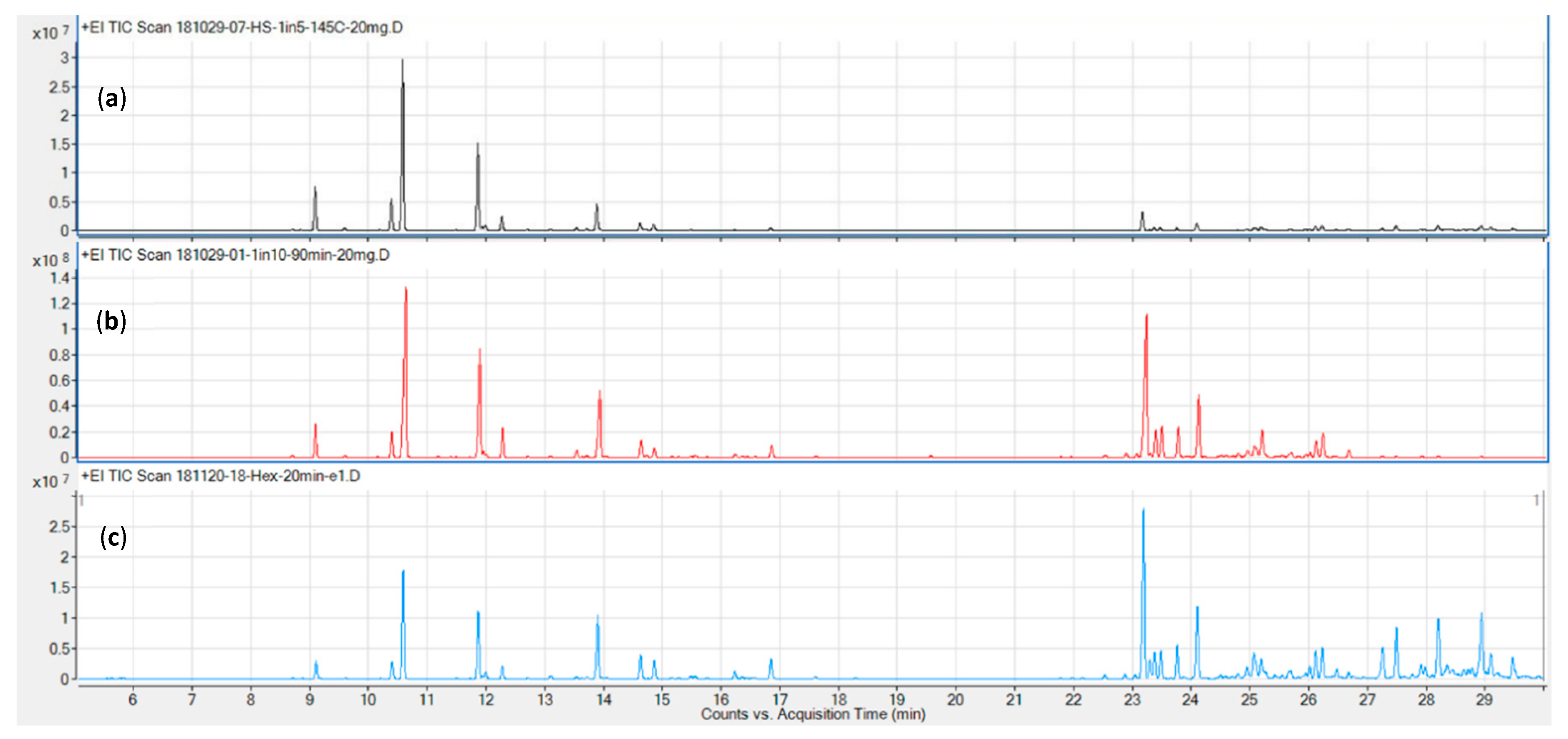
4.1. Headspace Techniques
4.2. Liquid Extracts
4.3. Standard Preparation
4.4. GC–MS Analysis
4.5. Validation Parameters
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Russo, E.B. The case for the entourage effect and conventional breeding of clinical cannabis No strain no gain. Front. Plant. Sci. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. History of Cannabis and Its Preparations in Saga, Science, and Sobriquet. Chem. Biodivers. 2007, 4, 1614–1648. [Google Scholar] [CrossRef]
- Wright, R. Cannabis Industry Report. Available online: https://www.everblucapital.com/wp-content/uploads/2017/11/EverBlu-Research-Cannabis-Industry-Report.pdf (accessed on 12 January 2019).
- Whiting, P.F.; Wolff, R.F.; Deshpande, S.; Di Nisio, M.; Duffy, S.; Hernandez, A.V.; Keurentjes, J.C.; Lang, S.; Misso, K.; Ryder, S.; et al. Cannabinoids for Medical Use: A Systematic Review and Meta-analysis. JAMA 2015, 313, 2456–2473. [Google Scholar] [CrossRef]
- Klumpers, L.E.; Thacker, D.L. A brief background on cannabis: From plant to medical indications. J. AOAC Int. 2019, 102, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.K.; Bohlmann, J. Terpenes in Cannabis sativa—From plant genome to humans. Plant. Sci. 2019, 284, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, F.A.; da Silva, L.R.; Coan, A.C. Potential Clinical Benefits of CBD-Rich Cannabis Extracts Over Purified CBD in Treatment-Resistant Epilepsy: Observational Data Meta-analysis. Front. Neurol. 2018, 9, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, J.S.; Downey, M.O.; Robinson, S.P. Expression of Vitis vinifera BANYULS modulates anthocyanin and tannin accumulation in tobacco (Tobaccum nicotiana). Plant. Physiol. in prep.
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef]
- Katrina, W.-G. The United Chemicals of Cannabis: Beneficial Effects of Cannabis Phytochemicals on the Brain and Cognition. In Recent Advances in Cannabinoid Research; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Baron, E.P. Medicinal Properties of Cannabinoids, Terpenes, and Flavonoids in Cannabis, and Benefits in Migraine, Headache, and Pain: An Update on Current Evidence and Cannabis Science. Headache J. Head Face Pain 2018, 58, 1139–1186. [Google Scholar] [CrossRef]
- Rohleder, C.; Müller, J.K.; Lange, B.; Leweke, F.M. Cannabidiol as a potential new type of an antipsychotic. A critical review of the evidence. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Bonn-Miller, M.O.; ElSohly, M.A.; Loflin, M.J.E.; Chandra, S.; Vandrey, R. Cannabis and cannabinoid drug development: Evaluating botanical versus single molecule approaches. Int. Rev. Psychiatry 2018, 30, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shabat, S.; Fride, E.; Sheskin, T.; Tamiri, T.; Rhee, M.H.; Vogel, Z.; Bisogno, T.; De Petrocellis, L.; Di Marzo, V.; Mechoulam, R. An entourage effect: Inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur. J. Pharmacol. 1998, 353, 23–31. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S. From gan-zi-gun-nu to anandamide and 2-arachidonoylglycerol: The ongoing story of cannabis. Nat. Prod. Rep. 1999, 16, 131–143. [Google Scholar] [CrossRef]
- McPartland, J.M.; Russo, E.B. Cannabis and Cannabis Extracts. J. Cannabis Ther. 2001, 1, 103–132. [Google Scholar] [CrossRef]
- Santiago, M.; Sachdev, S.; Arnold, J.; McGregor, I.; Connor, M. Absence of entourage: Terpenoids commonly found in Cannabis sativa do not modulate the functional activity of ∆9-THC at human CB1 and CB2 receptors. Cannabis Cannabinoid Res. 2019, 4, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef] [PubMed]
- Kamal, B.S.; Kamal, F.; Lantela, D. Cannabis and the Anxiety of Fragmentation—A Systems Approach for Finding an Anxiolytic Cannabis Chemotype. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Adams, T.B.; Taylor, S.V. Safety evaluation of essential oils: A constituent-based approach. In Handbook of Essential Oils: Science, Technology, and Applications; CRC Press: Boca Raton, FL, USA, 2010; pp. 185–208. [Google Scholar]
- Nuutinen, T. Medicinal properties of terpenes found in Cannabis sativa and Humulus lupulus. Eur. J. Med. Chem. 2018, 157, 198–228. [Google Scholar] [CrossRef]
- Do Vale, T.G.; Furtado, E.C.; Santos, J.G., Jr.; Viana, G.S.B. Central effects of citral, myrcene and limonene, constituents of essential oil chemotypes from Lippia alba (mill.) N.E. Brown. Phytomedicine 2002, 9, 709–714. [Google Scholar] [CrossRef]
- Leghissa, A.; Hildenbrand, Z.L.; Schug, K.A. A review of methods for the chemical characterization of cannabis natural products. J. Sep. Sci. 2018, 41, 398–415. [Google Scholar] [CrossRef]
- Giese, M.W.; Lewis, M.A.; Giese, L.; Smith, K.M. Development and Validation of a Reliable and Robust Method for the Analysis of Cannabinoids and Terpenes in Cannabis. J. AOAC Int. 2015, 98, 1503–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, E.A.; Wang, M.; Radwan, M.M.; Wanas, A.S.; Majumdar, C.G.; Avula, B.; Wang, Y.-H.; Khan, I.A.; Chandra, S.; Lata, H.; et al. Analysis of Terpenes in Cannabis sativa L. Using GC/MS: Method Development, Validation, and Application. Planta Med. 2019, 85, 431–438. [Google Scholar] [CrossRef]
- Fischedick, J.T.; Hazekamp, A.; Erkelens, T.; Choi, Y.H.; Verpoorte, R. Metabolic fingerprinting of Cannabis sativa L, cannabinoids and terpenoids for chemotaxonomic and drug standardization purposes. Phytochemistry 2010, 71, 2058–2073. [Google Scholar] [CrossRef]
- Fischedick, J.T. Identification of Terpenoid Chemotypes Among High (−)-trans-Delta9- Tetrahydrocannabinol-Producing Cannabis sativa L. Cultivars. Cannabis Cannabinoid Res. 2017, 2, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, B.; Ettre, L.S. Static Headspace–Gas. Chromatography: Theory and Practice, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Ascrizzi, R.; Flamini, G.; Giusiani, M.; Stefanelli, F.; Deriu, V.; Chericoni, S. VOCs as fingerprints for the chemical profiling of hashish samples analyzed by HS-SPME/GC–MS and multivariate statistical tools. Forensic Toxicol. 2018, 36, 243–260. [Google Scholar] [CrossRef]
- Rice, S.; Koziel, J.A. Characterizing the Smell of Marijuana by Odor Impact of Volatile Compounds: An Application of Simultaneous Chemical and Sensory Analysis. PLoS ONE 2015, 10, e0144160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvi, L.; Pentimalli, D.; Panseri, S.; Giupponi, L.; Gelmini, F.; Beretta, G.; Vitali, D.; Bruno, M.; Zilio, E.; Pavlovic, R.; et al. Comprehensive quality evaluation of medical Cannabis sativa L. inflorescence and macerated oils based on HS-SPME coupled to GC-MS and LC-HRMS (q-exactive orbitrap (R)) approach. J. Pharm. Biomed. Anal. 2018, 150, 208–219. [Google Scholar] [CrossRef]
- Shapira, A.; Berman, P.; Futoran, K.; Guberman, O.; Meiri, D. Tandem mass spectrometric quantification of 93 terpenoids in Cannabis using static headspace (SHS) injections. Anal. Chem. 2019, 91. [Google Scholar] [CrossRef] [Green Version]
- Snow, N.H.; Bullock, G.P. Novel techniques for enhancing sensitivity in static headspace extraction-gas chromatography. J. Chromatogr. A 2010, 1217, 2726–2735. [Google Scholar] [CrossRef]
- Leghissa, A. Method Development for Qualification and Quantification of Cannabinoids and Terpenes in Extracts by Gas Chromatography-Mass Spectrometry. Master’s Thesis, The University of Texas, Austin, TX, USA, August 2016. [Google Scholar]
- Leghissa, A.; Hildenbrand, Z.L.; Foss, F.W.; Schug, K.A. Determination of cannabinoids from a surrogate hops matrix using multiple reaction monitoring gas chromatography with triple quadrupole mass spectrometry. J. Sep. Sci. 2018, 41, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Van Den Dool, H.; Dec Kratz, P. A generalization of the retention index system including linear temperature programmed gas—Liquid partition chromatography. J. Chromatogr. A 1963, 11, 463–471. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
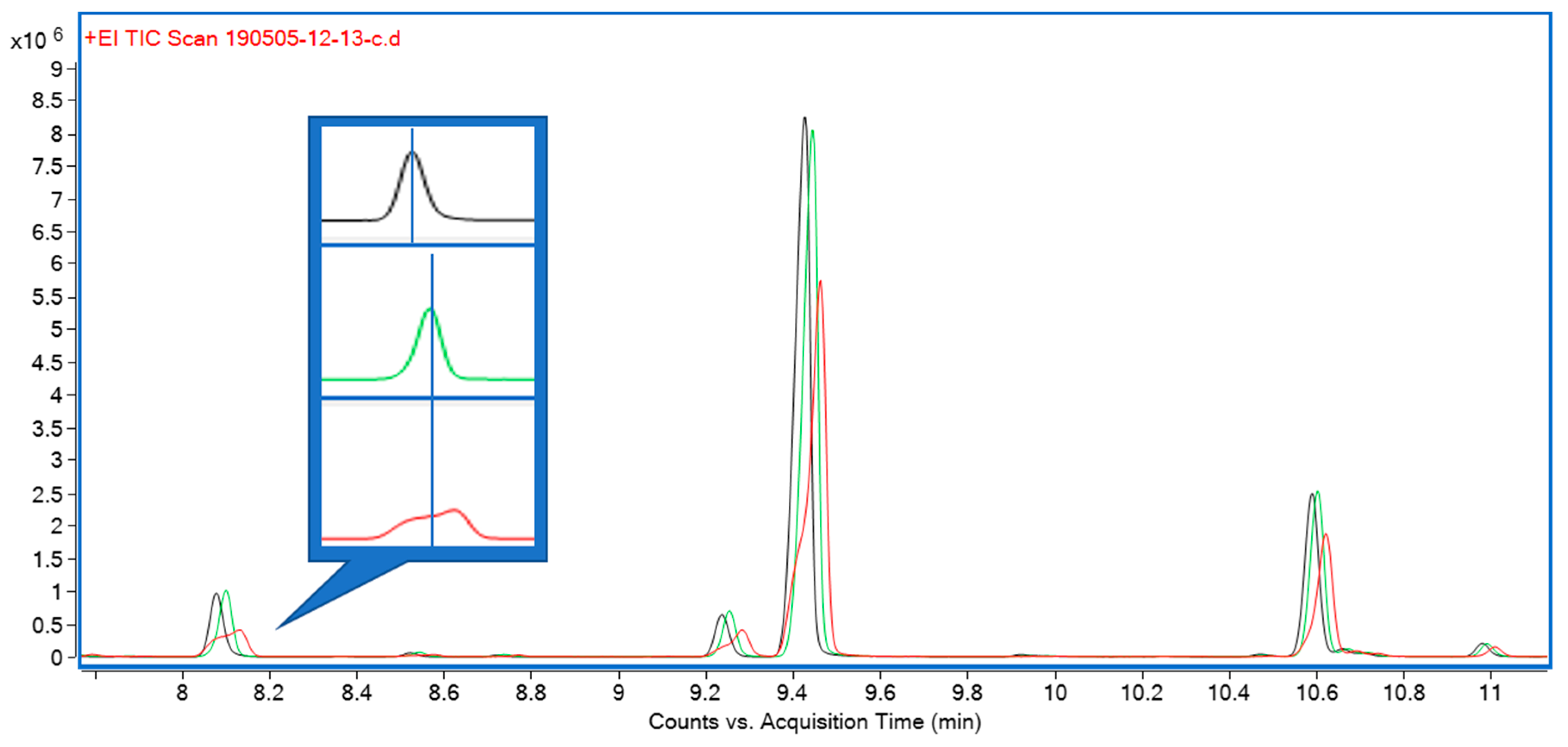{kind=link}
| Name | m/z | RT [Min] | RI | Rp | Rf | Status |
|---|---|---|---|---|---|---|
| α Thujene | 93.0 | 7.866 | 932 | n/a | 1.7 | RI |
| α-Pinene (+/−) | 93.0 | 8.056 | 940 | 1.9 | 1.6 | Val |
| Camphene | 93.0 | 8.505 | 958 | 4.8 | 1.7 | Val |
| Sabinene | 68.1 | 9.067 | 979 | 1.5 | 1.2 | Val |
| β-Pinene (+/−) | 69.2 | 9.215 | 985 | 1.3 | 1.4 | Val |
| Myrcene | 93.0 | 9.380 | 991 | 2.6 | 1.4 | Val |
| 3-Carene | 79.0 | 10.007 | 1014 | 1.9 | 1.6 | Val |
| α-Terpinene | 119.0 | 10.230 | 1023 | 2.0 | 1.6 | Val |
| p-Cymene | 93.0 | 10.437 | 1031 | 4.1 | 1.9 | Val |
| Limonene | 121.0 | 10.565 | 1035 | 3.0 | 15.7 | Val |
| β-Phellandrene | 93.1 | 10.660 | 1039 | 1.2 | 1 | specID, RI |
| Eucalyptol | 93.0 | 10.678 | 1040 | 1.7 | 2.1 | Val |
| Ocimene isomer | 105.1 | 10.960 | 1050 | 1.8 | 2.2 | Val |
| γ-Terpinene | 93.1 | 11.367 | 1064 | 2.9 | 2.3 | Val |
| 4-Thujanol | 93.1 | 11.750 | 1077 | 3.4 | 2.9 | specID, RI |
| Terpinolene | 93.0 | 12.150 | 1090 | 1.7 | 2.5 | Val |
| Fenchone | 81.1 | 12.333 | 1096 | 1.8 | 1.7 | confirmed |
| Linalool | 81.0 | 12.496 | 1101 | 1.4 | 2.7 | Val |
| Fenchol | 81.1 | 13.201 | 1128 | 7.4 | 2.0 | specID |
| trans-2-Pinanol | 93.1 | 13.421 | 1135 | 2.0 | 13 | specID |
| Isopulegol | 93.0 | 14.022 | 1156 | 1.6 | 2.8 | Val |
| Borneol | 95.1 | 14.758 | 1181 | 5.4 | 4.5 | confirmed |
| Dodecane | 57.1 | 15.370 | 1201 | 6.7 | 7.2 | IS |
| Nerolidol | 93.1 | 16.783 | 1253 | 2.6 | 22.5 | Val |
| β-Bergamotene | 119.1 | 21.015 | 1406 | 3.0 | 3.4 | specID |
| α-Bergamotene | 93.1 | 21.341 | 1420 | 3.6 | 2.3 | specID |
| trans-Caryophyllene | 93.1 | 21.600 | 1430 | 2.9 | 3.4 | Val |
| g-Elemene iso1 | 121.0 | 21.768 | 1437 | 1.4 | 1.8 | specID |
| Bergamotene iso3 | 119.0 | 21.848 | 1440 | 0.8 | 3.5 | specID, RI |
| α-Guaiene | 105.0 | 21.944 | 1444 | 1.7 | 1.8 | specID |
| Farnesene | 69.2 | 22.270 | 1456 | 4.1 | 2.1 | confirmed |
| Humulene | 69.1 | 22.524 | 1466 | 3.9 | 32.1 | Val |
| epi-β-Selinene | 93.1 | 23.396 | 1499 | 1.4 | 1.5 | specID |
| (−)-α-Selinene | 105.1 | 23.567 | 1506 | 1.7 | 1.1 | specID, RI |
| Sesquiterpenes, coeluting | 93.1 | 23.574 | 1506 | 1.5 | 1.4 | specID |
| δ-Guaiene | 161.0 | 23.662 | 1510 | 1.4 | 2.6 | specID, RI |
| β-Guaiene | 161.0 | 24.139 | 1530 | 1.2 | 1.5 | specID |
| Farnesol | 69.0 | 24.222 | 1534 | 3.2 | 3.7 | confirmed |
| α-Bisabolene | 93.1 | 24.512 | 1546 | 1.1 | 0.7 | specID |
| Guaia-3,9-diene | 161.1 | 24.572 | 1548 | 1.5 | 1.6 | specID |
| 3,7(11)-Selinadiene | 161.1 | 24.680 | 1553 | 1.6 | 1.1 | specID |
| β-cis-Caryophyllene | 93.0 | 24.968 | 1564 | 2.5 | 4.1 | Val |
| γ-Elemene iso2 | 121.1 | 25.152 | 1572 | 1.4 | 2.8 | specID |
| Caryophyllene Oxide | 93.0 | 25.691 | 1593 | 1.6 | 4.8 | Val |
| Guaiol | 93.0 | 25.970 | 1605 | 2.3 | 7.0 | Val |
| β-Cadinene | 189.1 | 26.666 | 1636 | 1.9 | 1.3 | specID |
| γ-Gurjunene | 59.1 | 27.400 | 1667 | 1.8 | 1.5 | specID |
| Sesquiterpene | 107.0 | 27.578 | 1675 | 1.2 | 1.4 | specID |
| α-Bisabolol | 93.0 | 27.989 | 1692 | 1.9 | n/a | Val |
| Compound | S | LOD | LOQ |
|---|---|---|---|
| α-Pinene (+/−) | 0.039129 | 0.022 | 0.068 |
| Camphene | 0.023849 | 0.032 | 0.097 |
| Sabinene | 0.038856 | 0.026 | 0.079 |
| β-Pinene (+/−) | 0.047192 | 0.017 | 0.052 |
| Myrcene | 0.028807 | 0.047 | 0.142 |
| 3-Carene | 0.030862 | 0.030 | 0.091 |
| α-Terpinene | 0.021459 | 0.038 | 0.115 |
| p-Cymene | 0.078714 | 0.021 | 0.065 |
| Limonene | 0.017349 | 0.066 | 0.200 |
| Eucalyptol | 0.008849 | 0.115 | 0.348 |
| Ocimene | 0.015782 | 0.057 | 0.174 |
| γ-Terpinene | 0.032899 | 0.046 | 0.139 |
| Terpinolene | 0.018898 | 0.045 | 0.137 |
| Linalool | 0.013307 | 0.060 | 0.181 |
| Isopulegol | 0.005376 | 0.045 | 0.136 |
| trans-Caryophyllene | 0.010502 | 0.092 | 0.326 |
| Humulene | 0.032275 | 0.021 | 0.065 |
| β-cis-Caryophyllene | 0.005202 | 0.388 | 1.177 |
| Caryophyllene Oxide | 0.004599 | 0.252 | 0.764 |
| Guaiol | 0.008546 | 0.107 | 0.324 |
| α-Bisabolol | 0.010087 | 1.144 | 3.468 |
| % Spike Recovery | |||
|---|---|---|---|
| High | Mid | Low | |
| α-Pinene (+/−) | 106.4 | 108.2 | 97.5 |
| Camphene | 112.6 | 121.4 | 119.0 |
| Sabinene | 108.8 | 115.7 | 98.7 |
| β-Pinene (+/−) | 110.7 | 113.7 | 102.2 |
| Myrcene | 106.5 | 104.8 | 95.5 |
| 3-Carene | 112.4 | 118.7 | 115.7 |
| α-terpinene | 78.8 | 84.5 | 81.8 |
| p-Cymene | 112.2 | 115.6 | 116.5 |
| Limonene | 110.6 | 114.6 | 107.9 |
| Eucalyptol | 116.6 | 109.9 | 119.2 |
| Ocimene | 101 | 105.7 | 98.6 |
| γ-Terpinene | 104.6 | 108.9 | 101.6 |
| Terpinolene | 97.1 | 100.4 | 93.0 |
| Linalool | 113.6 | 114.5 | 105.5 |
| Isopulegol | 61.8 | 66.2 | 82.6 |
| trans-Caryophyllene | 93.5 | 99.1 | 89.7 |
| Humulene | 92.5 | 98.3 | 90.7 |
| β-cis-Caryophyllene | 62.0 | 72.9 | 87.7 |
| Caryophyllene Oxide | 69.0 | 76.3 | 84.8 |
| Guaiol | 102.6 | 104.1 | 93.3 |
| α-Bisabolol | 98.7 | 100.3 | 93.5 |
| A1 Average | A1 %RSD | A2 Average | A2 %RSD | Combined Average | Combined %RSD | |
|---|---|---|---|---|---|---|
| α-Pinene (+/−) | 0.132 | 8.74 | 0.123 | 3.48 | 0.127 | 7.54 |
| Camphene | 0.035 | 8.27 | 0.033 | 3.61 | 0.034 | 7.01 |
| Sabinene | 0.005 | 5.07 | 0.005 | 2.54 | 0.005 | 3.92 |
| β-Pinene (+/−) | 0.233 | 7.56 | 0.221 | 2.95 | 0.227 | 6.17 |
| Myrcene (1:5) | 1.38 | 2.07 | 1.318 | 4.43 | 1.349 | 4.92 |
| 3-Carene | n/d | |||||
| α-Terpinene | 0.005 | 16.81 | 0.005 | 7.86 | 0.005 | 13.86 |
| p-Cymene | n/d | |||||
| Limonene | 1.753 | 5.74 | 1.744 | 3.4 | 1.749 | 4.62 |
| Eucalyptol | 0.027 | 20.86 | 0.028 | 13.55 | 0.028 | 18.68 |
| Ocimene | n/d | |||||
| γ-Terpinene | 0.006 | 4.87 | 0.006 | 4.38 | 0.006 | 5.61 |
| Terpinolene | 0.015 | 10.26 | 0.014 | 3.73 | 0.014 | 7.23 |
| Linalool | 0.017 | 2.77 | 0.018 | 2.75 | 0.017 | 4.46 |
| Isopulegol | 0.06 | 2.43 | 0.06 | 1.97 | 0.06 | 2.52 |
| Nerolidol | n/d | |||||
| trans-Caryophyllene | 1.492 | 1.76 | 1.449 | 1.93 | 1.47 | 2.31 |
| Humulene | 0.419 | 2.44 | 0.408 | 1.66 | 0.413 | 2.6 |
| β-cis-Caryophyllene | 0.198 | 1.44 | 0.201 | 1.9 | 0.2 | 1.65 |
| Caryophyllene oxide | 0.121 | 2.79 | 0.129 | 2.11 | 0.125 | 3.8 |
| Guaiol | 0.479 | 1.86 | 0.491 | 2.13 | 0.485 | 2.57 |
| α-Bisabolol | 0.553 | 1.13 | 0.565 | 1.65 | 0.559 | 2.62 |
| Name | m/z | RT | Source |
|---|---|---|---|
| α-Pinene (+/−) | 93.0 | 8.056 | CT1 |
| Camphene | 93.0 | 8.505 | CT1 |
| Sabinene | 93.0 | 9.067 | Ind. |
| β-Pinene (+/−) | 93.0 | 9.215 | CT1 |
| Myrcene | 93.0 | 9.380 | CT1 |
| 3-Carene | 93.0 | 10.007 | CT1 |
| α-terpinene | 93.0 | 10.230 | CT1 |
| p-Cymene | 119.0 | 10.437 | CT1 |
| Limonene | 68.1 | 10.565 | CT1 |
| Eucalyptol | 81.0 | 10.678 | CT2 |
| Ocimene isomer | 93.1 | 10.960 | CT1 |
| γ-Terpinene | 93.1 | 11.367 | CT1 |
| Terpinolene | 93.1 | 12.150 | CT1 |
| Linalool | 93.0 | 12.496 | CT1 |
| Isopulegol | 121.0 | 14.022 | CT1 |
| Nerolidol | 69.1 | 16.783 | CT1 |
| trans-Caryophyllene | 93.0 | 21.600 | CT1 |
| Humulene | 93.0 | 22.524 | CT1 |
| β-cis-Caryophyllene | 69.2 | 24.968 | CT1 |
| Caryophyllene Oxide | 79.0 | 25.691 | CT2 |
| Guaiol | 105.1 | 25.970 | CT1 |
| α-Bisabolol | 93.0 | 27.989 | CT1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krill, C.; Rochfort, S.; Spangenberg, G. A High-Throughput Method for the Comprehensive Analysis of Terpenes and Terpenoids in Medicinal Cannabis Biomass. Metabolites 2020, 10, 276. https://doi.org/10.3390/metabo10070276
Krill C, Rochfort S, Spangenberg G. A High-Throughput Method for the Comprehensive Analysis of Terpenes and Terpenoids in Medicinal Cannabis Biomass. Metabolites. 2020; 10(7):276. https://doi.org/10.3390/metabo10070276
Chicago/Turabian StyleKrill, Christian, Simone Rochfort, and German Spangenberg. 2020. "A High-Throughput Method for the Comprehensive Analysis of Terpenes and Terpenoids in Medicinal Cannabis Biomass" Metabolites 10, no. 7: 276. https://doi.org/10.3390/metabo10070276
APA StyleKrill, C., Rochfort, S., & Spangenberg, G. (2020). A High-Throughput Method for the Comprehensive Analysis of Terpenes and Terpenoids in Medicinal Cannabis Biomass. Metabolites, 10(7), 276. https://doi.org/10.3390/metabo10070276