Comprehensive Vitamer Profiling of Folate Mono- and Polyglutamates in Baker’s Yeast (Saccharomyces cerevisiae) as a Function of Different Sample Preparation Procedures

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
2.1. Optimization of Sample Preparation Procedures for the Investigation of Folate Metabolites by UHPLC-Q-ToF-MS
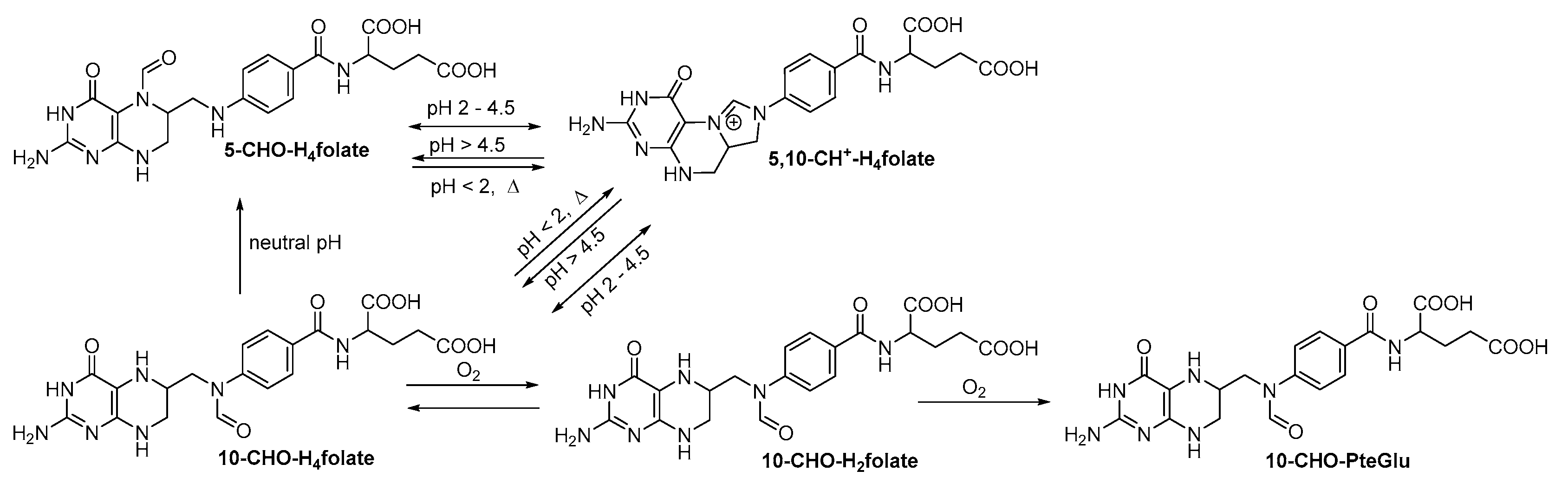
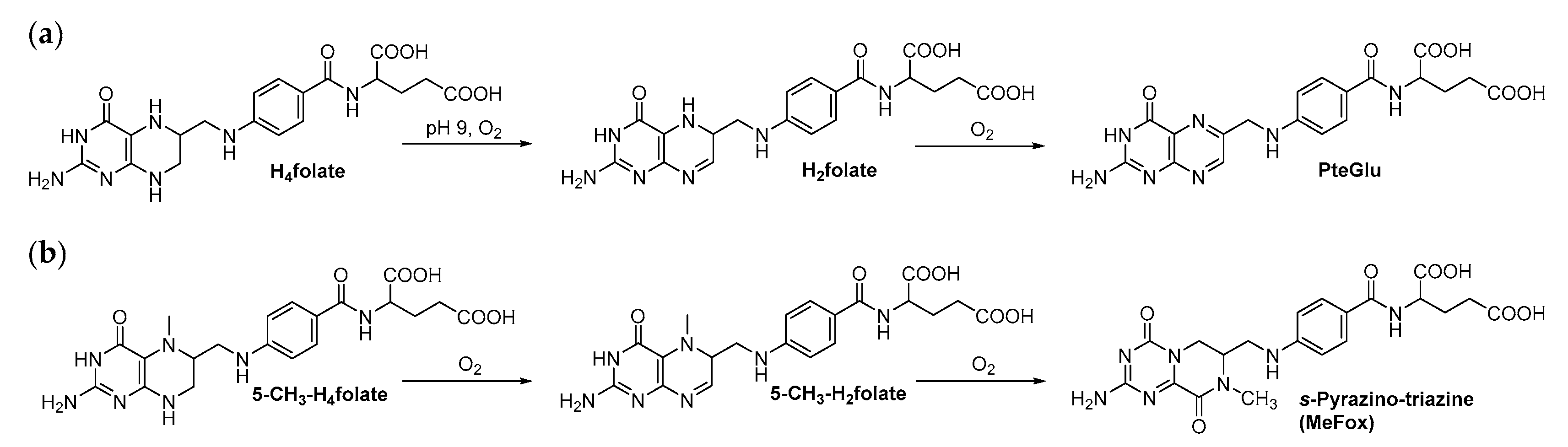
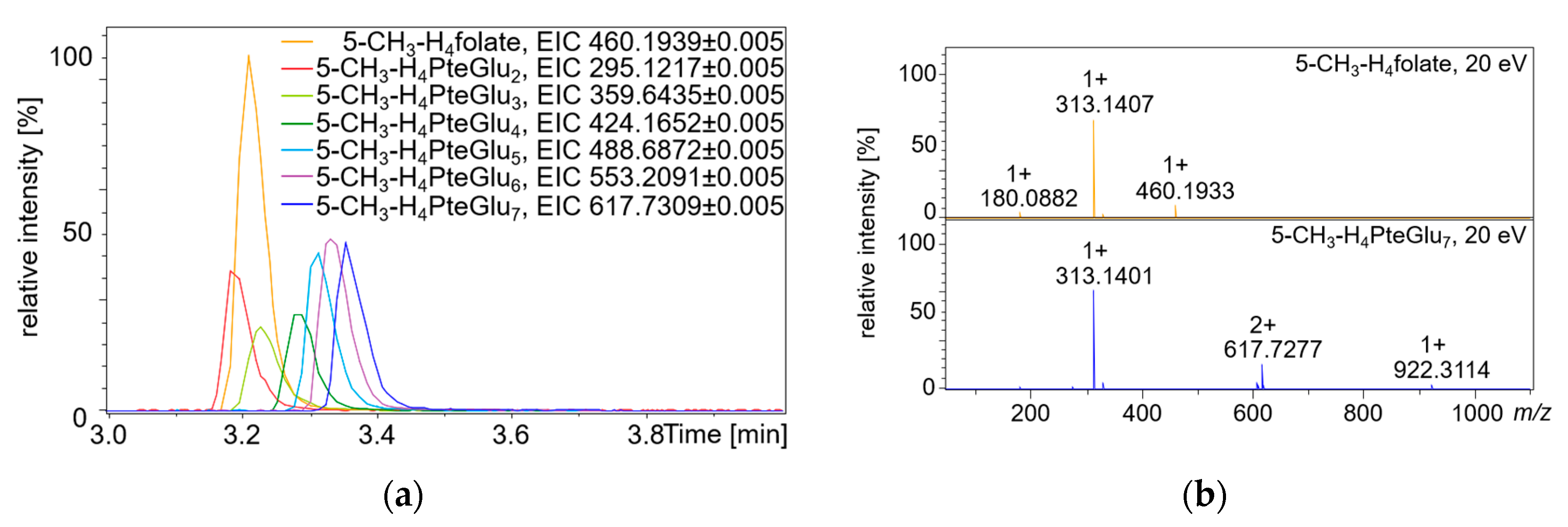
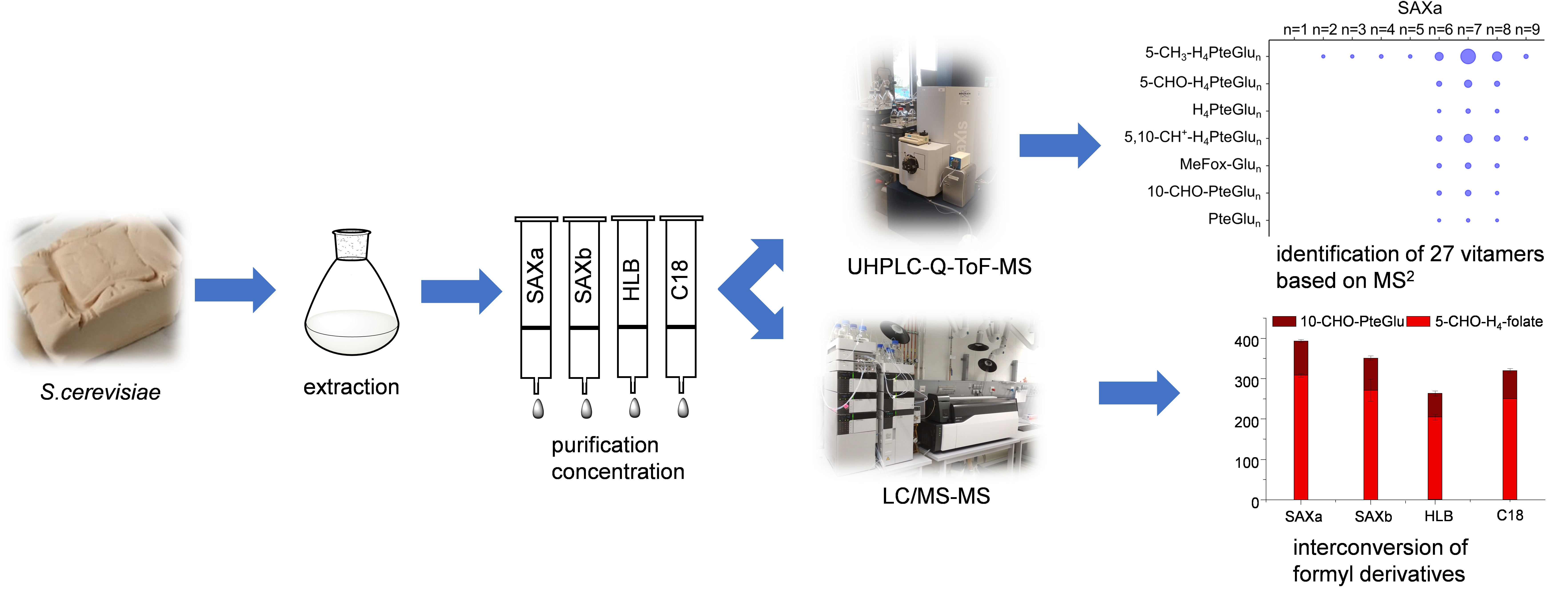
2.2. Qualitative Profiling of Folate Metabolites by UHPLC-Q-ToF-MS
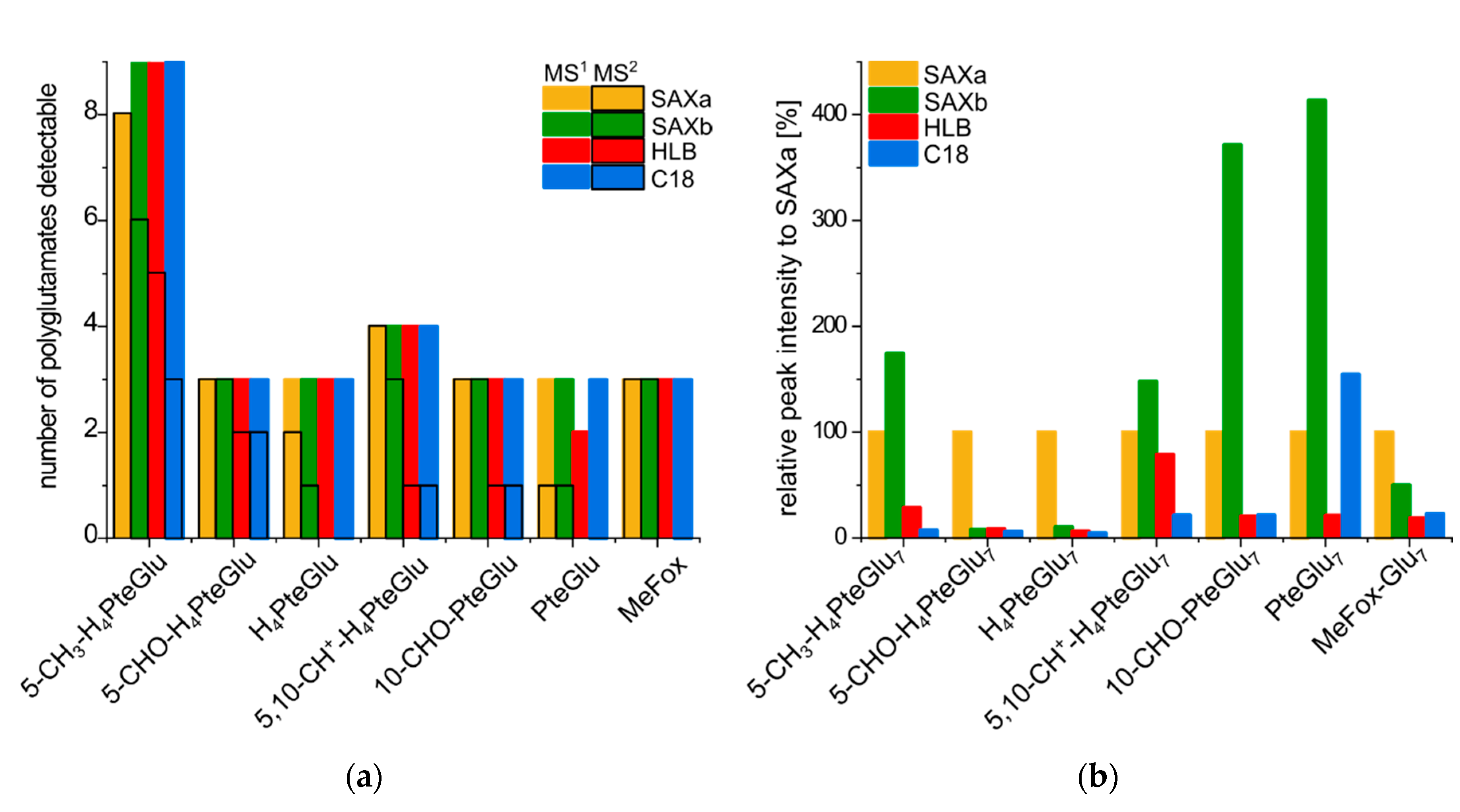
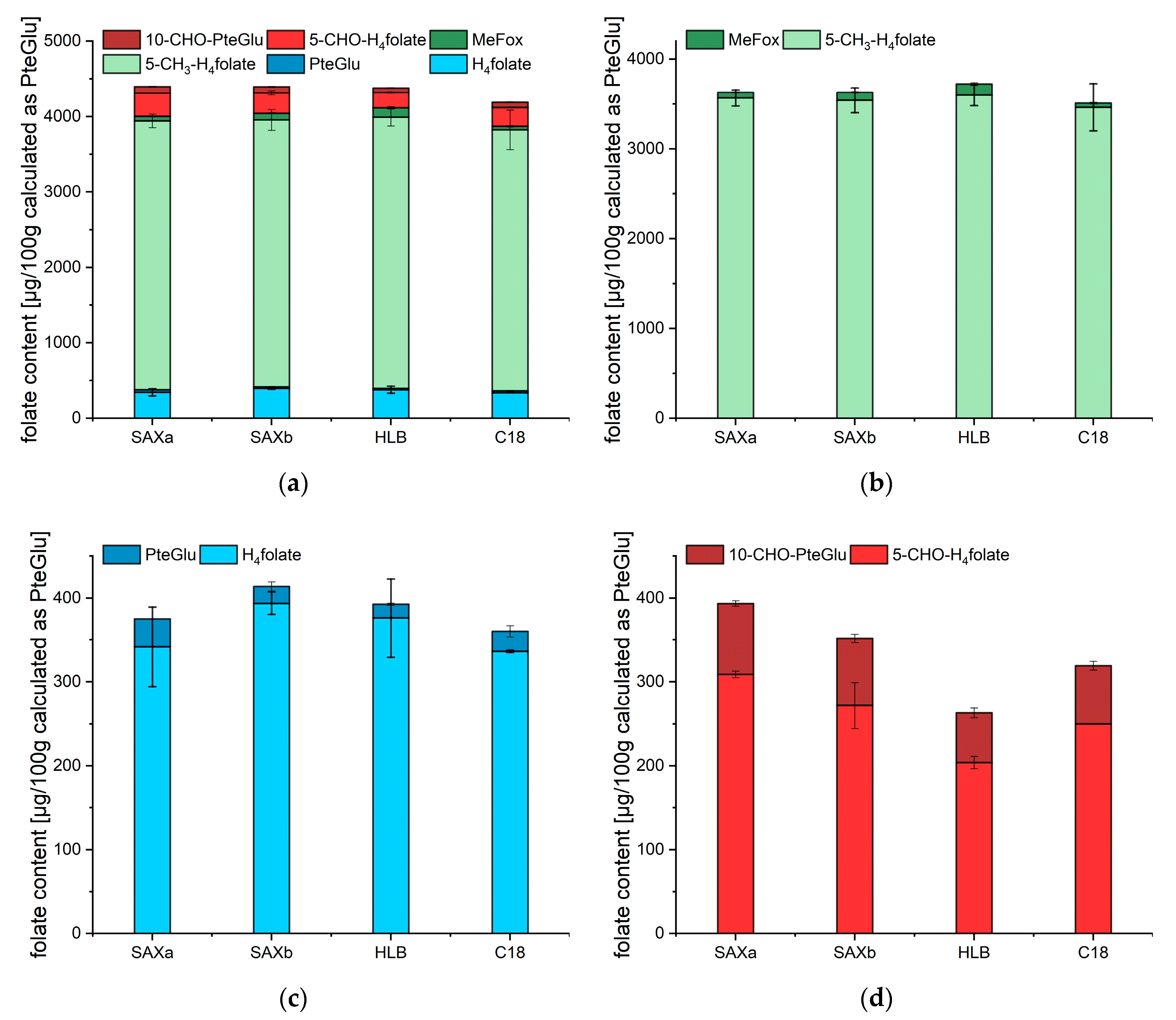
2.3. Quantitative Analysis of the Total Folate Content after Enzymatic Deconjugation and Purification with Different SPE Materials
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Solutions for Sample Extraction and Solid Phase Extraction Procedures
4.3. Sample Collection
4.4. Sample Extraction Procedure for the Analysis of the Total Folate Content
4.5. Sample Extraction Procedure for the Analysis of the Pteroylpolyglutamates
4.6. SPE-Procedure for Sample Extracts with SAX Cartridges (Procedures SAXa and SAXb)
4.7. SPE-Procedure for Sample Extracts with HLB Cartridges
4.8. SPE-Procedure for Sample Extracts with C18 Cartridges
4.9. Determination of Folate Standard Concentrations by High Performance Liquid Chromatography Diode Array Detector (HPLC-DAD)
4.10. Determination of the Total Folate Content by LC-MS/MS
4.11. Synthesis of 5-CH3-H4PteGlun and MeFox-PteGlu7
4.12. Optimization of the Analysis of the Folate Metabolite Profile by UHPLC-Q-ToF-MS
4.13. Data Processing
5. Conclusions
- Relatively low concentration of analytes (µM range);
- Insufficient folate extraction in typical un-targeted set-ups (ice-cold extraction);
- Insufficient dissolution of folates in the organic solvent mixtures usually used for sample preparation;
- Insufficient optimization of (SPE) purification procedures;
- Loss of intensity for the more general SPE procedures applied (RP phase and polymeric resin);
- Low ionizing potential using 0.1% formic acid concentration in the mobile phase of UHPLC separation coupled to MS analysis;
- Interconversion and oxidation reactions need to be taken into account.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef]
- Fiehn, O.; Weckwerth, W. Deciphering metabolic networks. Eur. J. Biochem. 2003, 270, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuckovic, D. Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 1523–1548. [Google Scholar] [CrossRef]
- Rahman, M.; Hasan, M.R. Pentose Phosphate Pathway in Disease and Therapy. Adv. Mater. Res. 2014, 995, 1–27. [Google Scholar] [CrossRef]
- Garavito, M.F.; Narvaez-Ortiz, H.Y.; Zimmermann, B.H. Pyrimidine Metabolism: Dynamic and Versatile Pathways in Pathogens and Cellular Development. J. Genet. Genom. 2015, 42, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.I.; Dunn, W.B.; Griffin, J.L.; Allwood, J.W.; Goodacre, R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics 2007, 8, 1243–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowda, G.A.; Zhang, S.; Gu, H.; Asiago, V.; Shanaiah, N.; Raftery, D. Metabolomics-based methods for early disease diagnostics. Expert Rev. Mol. Diagn. 2008, 8, 617–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Fanos, V.; Antonucci, R.; Atzori, L. Metabolomics in the developing infant. Curr. Opin. Pediatr. 2013, 25, 604–611. [Google Scholar] [CrossRef]
- Pereira, S.; Kildegaard, H.F.; Andersen, M.R. Impact of CHO Metabolism on Cell Growth and Protein Production: An Overview of Toxic and Inhibiting Metabolites and Nutrients. Biotechnol. J. 2018, 13, e1700499. [Google Scholar] [CrossRef] [Green Version]
- Shulaev, V.; Cortes, D.; Miller, G.; Mittler, R. Metabolomics for plant stress response. Physiol. Plant. 2008, 132, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lankadurai, B.P. Environmental metabolomics: An emerging approach to study organism responses to environmental stressors. Environ. Rev. 2013, 21, 180–205. [Google Scholar] [CrossRef]
- Parihar, P.; Singh, S.; Singh, R.; Singh, V.P.; Prasad, S.M. Effect of salinity stress on plants and its tolerance strategies: A review. Environ. Sci. Pollut. Res. 2015, 22, 4056–4075. [Google Scholar] [CrossRef]
- Castrillo, J.I.; Hayes, A.; Mohammed, S.; Gaskell, S.J.; Oliver, S.G. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry 2003, 62, 929–937. [Google Scholar] [CrossRef]
- Crutchfield, C.A.; Lu, W.; Melamud, E.; Rabinowitz, J.D. Mass spectrometry-based metabolomics of yeast. Methods Enzymol. 2010, 470, 393–426. [Google Scholar] [CrossRef]
- Goncerzewicz, A.; Misiewicz, A. The sequence diversity and expression among genes of the folic acid biosynthesis pathway in industrial Saccharomyces strains. Acta. Biochim. Pol. 2015, 62, 841–850. [Google Scholar] [CrossRef]
- Álvarez-Sánchez, B.; Priego-Capote, F.; Castro, M.D.L.d. Metabolomics analysis II. Preparation of biological samples prior to detection. Trends Anal. Chem. 2010, 29, 120–127. [Google Scholar] [CrossRef]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for metabolomics: Access to the metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef]
- Ramirez-Gaona, M.; Marcu, A.; Pon, A.; Guo, A.C.; Sajed, T.; Wishart, N.A.; Karu, N.; Feunang, Y.D.; Arndt, D.; Wishart, D.S. YMDB 2.0: A significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [Google Scholar] [CrossRef]
- Saini, R.K.; Nile, S.H.; Keum, Y.-S. Folates: Chemistry, analysis, occurrence, biofortification and bioavailability. Food Res. Int. 2016, 89, 1–13. [Google Scholar] [CrossRef]
- Finglas, P.M.; Wright, A.J.A. Folate bioavailability and health. Phytochem. Rev. 2002, 1, 189–198. [Google Scholar] [CrossRef]
- Shane, B. Folate Chemistry and Metabolism. In Folate in Health and Disease, 2nd ed.; Bailey, L.B., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2010; pp. 1–24. [Google Scholar]
- Zheng, Y.; Cantley, L.C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 2018, 216, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Schirch, V.; Strong, W.B. Interaction of folylpolyglutamates with enzymes in one-carbon metabolism. Arch. Biochem. Biophys. 1989, 269, 371–380. [Google Scholar] [CrossRef]
- Appling, D.R. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991, 5, 2645–2651. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Iyer, R.; Tomar, S.K. Folate: A functional food constituent. J. Food Sci. 2009, 74, R114–R122. [Google Scholar] [CrossRef]
- Shane, B. Folate and vitamin B12 metabolism: Overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food Nutr. Bull. 2008, 29, discussion S17–S19. [Google Scholar] [CrossRef]
- Duthie, S.J. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. J. Inherit. Metab. Dis. 2011, 34, 101–109. [Google Scholar] [CrossRef]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [Green Version]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef]
- Cook, J.W.; Taylor, L.M.; Orloff, S.L.; Landry, G.J.; Moneta, G.L.; Porter, J.M. Homocysteine and arterial disease. Experimental mechanisms. Vasc. Pharmcol. 2002, 38, 293–300. [Google Scholar] [CrossRef]
- McCully, K.S. Homocysteine, vitamins, and prevention of vascular disease. Mil. Med. 2004, 169, 325–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.; Homocysteine, B. Vitamins, and risk of cardiovascular disease. Heart 2000, 83, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansky, L.V.; Andermann, E.; Rosenblatt, D.; Sherwin, A.L.; Andermann, F. Anticonvulsants, folate levels, and pregnancy outcome: A prospective study. Ann. Neurol. 1987, 21, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.D.; Gregory, J.F. Folate biosynthesis, turnover, and transport in plants. Annu. Rev. Plant Biol. 2011, 62, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Bailey, L.B.; Berry, R.J. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients 2011, 3, 370–384. [Google Scholar] [CrossRef] [Green Version]
- Canfield, M.A.; Collins, J.S.; Botto, L.D.; Williams, L.J.; Mai, C.T.; Kirby, R.S.; Pearson, K.; Devine, O.; Mulinare, J. Changes in the birth prevalence of selected birth defects after grain fortification with folic acid in the United States: Findings from a multi-state population-based study. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 679–689. [Google Scholar] [CrossRef]
- Honein, M.A.; Paulozzi, L.J.; Mathews, T.J.; Erickson, J.D.; Wong, L.-Y.C. Impact of Folic Acid Fortification of the US Food Supply on the Occurrence of Neural Tube Defects. JAMA 2001, 285, 2981–2986. [Google Scholar] [CrossRef] [Green Version]
- Titcomb, T.J.; Tanumihardjo, S.A. Global Concerns with B Vitamin Statuses: Biofortification, Fortification, Hidden Hunger, Interactions, and Toxicity. Compr. Rev. Food Sci. Food. Saf. 2019, 18, 1968–1984. [Google Scholar] [CrossRef] [Green Version]
- Wien, T.N.; Pike, E.; Wisløff, T.; Staff, A.; Smeland, S.; Klemp, M. Cancer risk with folic acid supplements: A systematic review and meta-analysis. BMJ Open 2012, 2, e000653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancquaert, D.; Storozhenko, S.; Van Daele, J.; Stove, C.; Visser, R.G.; Lambert, W.; Van Der Straeten, D. Enhancing pterin and para-aminobenzoate content is not sufficient to successfully biofortify potato tubers and Arabidopsis thaliana plants with folate. J. Exp. Bot. 2013, 64, 3899–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Diaz de Garza, R.; Quinlivan, E.P.; Klaus, S.M.J.; Basset, G.J.C.; Gregory, J.F.r.; Hanson, A.D. Folate biofortification in tomatoes by engineering the pteridine branch of folate synthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 13720–13725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelova, V.; Ambach, L.; Rébeillé, F.; Stove, C.; van der Straeten, D. Folates in Plants: Research Advances and Progress in Crop Biofortification. Front. Chem. 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, T.; Rosenberg, I.; Selhub, J.; Kishore, G.; Beachy, R.; Schubert, K. Enhancement of folates in plants through metabolic engineering. Proc. Natl. Acad. Sci. USA 2004, 101, 5158–5163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Walkey, C.J.; Green, T.J.; van Vuuren, H.J.; Kitts, D.D. Enhancing the natural folate level in wine using bioengineering and stabilization strategies. Food Chem. 2016, 194, 26–31. [Google Scholar] [CrossRef]
- Strobbe, S.; van der Straeten, D. Folate biofortification in food crops. Curr. Opin. Biotechnol. 2017, 44, 202–211. [Google Scholar] [CrossRef]
- Quinlivan, E.P.; Hanson, A.D.; Gregory, J.F. The analysis of folate and its metabolic precursors in biological samples. Anal. Biochem. 2006, 348, 163–184. [Google Scholar] [CrossRef]
- Rebeille, F.; Ravanel, S.; Jabrin, S.; Douce, R.; Storozhenko, S.; Van Der Straeten, D. Folates in plants: Biosynthesis, distribution, and enhancement. Physiol. Plant. 2006, 126, 330–342. [Google Scholar] [CrossRef]
- Gazzali, A.M.; Lobry, M.; Colombeau, L.; Acherar, S.; Azaïs, H.; Mordon, S.; Arnoux, P.; Baros, F.; Vanderesse, R.; Frochot, C. Stability of folic acid under several parameters. Eur. J. Pharm. Sci. 2016, 93, 419–430. [Google Scholar] [CrossRef]
- Delchier, N.; Ringling, C.; Cuvelier, M.-E.; Courtois, F.; Rychlik, M.; Renard, C.M.G.C. Thermal degradation of folates under varying oxygen conditions. Food Chem. 2014, 165, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Indrawati, I.; Van Loey, A.; Hendrickx, M. Pressure and temperature stability of 5-methyltetrahydrofolic acid: A kinetic study. J. Agric. Food Chem. 2005, 53, 3081–3087. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, V.; Zhang, G.-F.; Storozhenko, S.; van der Straeten, D.; Lambert, W.E. pH stability of individual folates during critical sample preparation steps in prevision of the analysis of plant folates. Phytochem. Anal. 2007, 18, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Lucock, M.D.; Green, M.; Hartley, R.; Levene, M.I. Physicochemical and biological factors influencing methylfolate stability: Use of dithiothreitol for HPLC analysis with electrochemical detection. Food Chem. 1993, 47, 79–86. [Google Scholar] [CrossRef]
- Indrawati; Arroqui, C.; Messagie, I.; Nguyen, M.T.; Van Loey, A.; Hendrickx, M. Comparative study on pressure and temperature stability of 5-methyltetrahydrofolic acid in model systems and in food products. J. Agric. Food Chem. 2004, 52, 485–492. [Google Scholar] [CrossRef]
- Patring, J.D.M.; Johansson, M.S.; Yazynina, E.; Jastrebova, J.A. Evaluation of impact of different antioxidants on stability of dietary folates during food sample preparation and storage of extracts prior to analysis. Anal. Chim. Acta 2005, 553, 36–42. [Google Scholar] [CrossRef]
- Baggott, J.E. Hydrolysis of 5,10-Methenyltetrahydrofolate to 5-Formyltetrahydrofolate at pH 2.5 to 4.5. Biochemistry 2000, 39, 14647–14653. [Google Scholar] [CrossRef]
- Jägerstad, M.; Jastrebova, J. Occurrence, stability, and determination of formyl folates in foods. J. Agric. Food Chem. 2013, 61, 9758–9768. [Google Scholar] [CrossRef]
- Fazili, Z.; Sternberg, M.R.; Paladugula, N.; Whitehead, R.D., Jr.; Chen, H.; Pfeiffer, C.M. The loss of 5-methyltetrahydrofolate in human serum under suboptimal preanalytical conditions can only partially be recovered by an oxidation product. J. Nutr. 2014, 144, 1873–1879. [Google Scholar] [CrossRef]
- Ringling, C.; Rychlik, M. Origins of the difference between food folate analysis results obtained by LC-MS/MS and microbiological assays. Anal. Bioanal. Chem. 2017, 409, 1815–1825. [Google Scholar] [CrossRef] [Green Version]
- Gapski, G.R.; Whiteley, J.M.; Huennekens, F.M. Hydroxylated derivatives of 5-methyl-5,6,7,8-tetrahydrofolate. Biochemistry 1971, 10, 2930–2934. [Google Scholar] [CrossRef] [PubMed]
- Arcot, J.; Shrestha, A. Folate: Methods of analysis. Trends Food Sci. Technol. 2005, 16, 253–266. [Google Scholar] [CrossRef]
- Garratt, L.C.; Ortori, C.A.; Tucker, G.A.; Sablitzky, F.; Bennett, M.J.; Barrett, D.A. Comprehensive metabolic profiling of mono- and polyglutamated folates and their precursors in plant and animal tissue using liquid chromatography/negative ion electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2390–2398. [Google Scholar] [CrossRef]
- Leung, K.-Y.; Castro, S.C.P.; Cabreiro, F.; Gustavsson, P.; Copp, A.J.; Greene, N.D.E. Folate metabolite profiling of different cell types and embryos suggests variation in folate one-carbon metabolism, including developmental changes in human embryonic brain. Mol. Cell. Biochem. 2013, 378, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Duan, H.; Zou, Y.; Qiu, R.; Wang, C. Quantification of Total Folate, Folate Species and Polyglutamyl Folate Distribution in Winged Beans (Psophocarus tetragonolobus (L) DC) from Different Cultivars and Growth Stages by Ultra-High Performance Liquid Chromatography Tandem Mass Spectrometry. J. Nutr. Sci. Vitamnol. 2017, 63, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Riedl, K.M.; Schwartz, S.J. A liquid chromatography-tandem mass spectrometric method for quantitative determination of native 5-methyltetrahydrofolate and its polyglutamyl derivatives in raw vegetables. J. Chromatogr. B 2010, 878, 2949–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Riedl, K.M.; Somerville, J.; Balasubramaniam, V.M.; Schwartz, S.J. Influence of high-pressure processing on the profile of polyglutamyl 5-methyltetrahydrofolate in selected vegetables. J. Agric. Food Chem. 2011, 59, 8709–8717. [Google Scholar] [CrossRef] [Green Version]
- Gregory, J.F. Bioavailability of folate. Eur. J. Clin. Nutr. 1997, 51, S54–S59. [Google Scholar]
- Patring, J.D.M.; Jastrebova, J.A.; Hjortmo, S.B.; Andlid, T.A.; Jägerstad, I.M. Development of a simplified method for the determination of folates in baker’s yeast by HPLC with ultraviolet and fluorescence detection. J. Agric. Food Chem. 2005, 53, 2406–2411. [Google Scholar] [CrossRef]
- Seyoum, E.; Selhub, J. Properties of Food Folates Determined by Stability and Susceptibility to Intestinal Pteroylpolyglutamate Hydrolase Action. J. Nutr. 1998, 128, 1956–1960. [Google Scholar] [CrossRef]
- Hjortmo, S.; Patring, J.; Jastrebova, J.; Andlid, T. Inherent biodiversity of folate content and composition in yeasts. Trends Food Sci. Technol. 2005, 16, 311–316. [Google Scholar] [CrossRef]
- Gregory, J.F. Chemical and nutritional aspects of folate research: Analytical procedures, methods of folate synthesis, stability, and bioavailability of dietary folates. Adv. Food Nutr. Res. 1989, 33, 1–101. [Google Scholar] [PubMed]
- Nilsson, C.; Johansson, M.; Yazynina, E.; Strlsj, L.; Jastrebova, J. Solid-phase extraction for HPLC analysis of dietary folates. Eur. Food Res. Technol. 2004, 219, 199–204. [Google Scholar] [CrossRef]
- Patring, J.D.M.; Jastrebova, J.A. Application of liquid chromatography-electrospray ionisation mass spectrometry for determination of dietary folates: Effects of buffer nature and mobile phase composition on sensitivity and selectivity. J. Chromatogr. A 2007, 1143, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Strandler, H.S.; Patring, J.; Jagerstad, M.; Jastrebova, J. Challenges in the determination of unsubstituted food folates: Impact of stabilities and conversions on analytical results. J. Agric. Food Chem. 2015, 63, 2367–2377. [Google Scholar] [CrossRef]
- Shohag, M.J.I.; Yang, Q.; Wei, Y.; Zhang, J.; Khan, F.Z.; Rychlik, M.; He, Z.; Yang, X. A rapid method for sensitive profiling of folates from plant leaf by ultra-performance liquid chromatography coupled to tandem quadrupole mass spectrometer. J. Chromatogr. B 2017, 1040, 169–179. [Google Scholar] [CrossRef]
- Rebello, T. Trace enrichment of biological folates on solid-phase adsorption cartridges and analysis by high-pressure liquid chromatography. Anal. Biochem. 1987, 166, 55–64. [Google Scholar] [CrossRef]
- Pawlosky, R.J.; Flanagan, V.P.; Pfeiffer, C.M. Determination of 5-methyltetrahydrofolic acid in human serum by stable-isotope dilution high-performance liquid chromatography-mass spectrometry. Anal. Biochem. 2001, 298, 299–305. [Google Scholar] [CrossRef]
- Chandra-Hioe, M.V.; Bucknall, M.P.; Arcot, J. Folic Acid-fortified Flour: Optimised and Fast Sample Preparation Coupled with a Validated High-Speed Mass Spectrometry Analysis Suitable for a Fortification Monitoring Program. Food Anal. Methods 2013, 6, 1416–1423. [Google Scholar] [CrossRef]
- Fazili, Z.; Whitehead, R.D.; Paladugula, N.; Pfeiffer, C.M. A high-throughput LC-MS/MS method suitable for population biomonitoring measures five serum folate vitamers and one oxidation product. Anal. Bioanal. Chem. 2013, 405, 4549–4560. [Google Scholar] [CrossRef] [Green Version]
- Camara, J.E.; Lowenthal, M.S.; Phinney, K.W. Determination of fortified and endogenous folates in food-based Standard Reference Materials by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 4561–4568. [Google Scholar] [CrossRef] [PubMed]
- Kiekens, F.; van Daele, J.; Blancquaert, D.; van der Straeten, D.; Lambert, W.E.; Stove, C.P. A validated ultra-high-performance liquid chromatography-tandem mass spectrometry method for the selective analysis of free and total folate in plasma and red blood cells. J. Chromatogr. A 2015, 1398, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, B.C.; Pfeiffer, C.M.; Margolis, S.A.; Nelson, C.P. Solid-phase extraction–electrospray ionization mass spectrometry for the quantification of folate in human plasma or serum. Anal. Biochem. 2004, 325, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kok, R.M.; Smith, D.E.C.; Dainty, J.R.; Akker, J.T.V.D.; Finglas, P.M.; Smulders, Y.M.; Jakobs, C.; Meer, K.D. 5-Methyltetrahydrofolic acid and folic acid measured in plasma with liquid chromatography tandem mass spectrometry: Applications to folate absorption and metabolism. Anal. Biochem. 2004, 326, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Rosebrock, A.P.; Caudy, A.A. Metabolite Extraction from Saccharomyces cerevisiae for Liquid Chromatography–Mass Spectrometry. Cold Spring Harb. Protoc. 2017, 2017, 089086. [Google Scholar] [CrossRef]
- Rychlik, M.; Asam, S. Stabilisotopenverdünnungsanalysen zur Quantifizierung organischer Spurenkomponenten in der Lebensmittelanalytik. UWSF 2009, 21, 470–482. [Google Scholar] [CrossRef] [Green Version]
- Villas-Boas, S.G.; Hojer-Pedersen, J.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef] [Green Version]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative Evaluation of Intracellular Metabolite Extraction Techniques for Yeast Metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef]
- de Koning, W.; van Dam, K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992, 204, 118–123. [Google Scholar] [CrossRef]
- Gonzalez, B.; Francois, J.; Renaud, M. A rapid and reliable method for metabolite extraction in yeast using boiling buffered ethanol. Yeast 1997, 13, 1347–1355. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.; Wohlgemuth, G.; Park, H.S.; Fiehn, O.; Kim, K. Evaluation and Optimization of Metabolome Sample Preparation Methods for Saccharomyces cerevisiae. Anal. Chem. 2013, 85, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Santiago, M.; Priego-Capote, F.; Castro, M. Enhancing detection coverage in untargeted metabolomics analysis by solid-phase extraction on-line coupled to LC-MS/MS. Electrophoresis 2015, 36, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Michopoulos, F.; Gika, H.; Palachanis, D.; Theodoridis, G.; Wilson, I.D. Solid phase extraction methodology for UPLC-MS based metabolic profiling of urine samples. Electrophoresis 2015, 36, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Schertel, M.E.; Boehne, J.W.; Libby, D.A. Folic Acid Derivatives in Yeast. J. Biol. Chem. 1965, 240, 3154–3158. [Google Scholar] [PubMed]
- Stokstad, E.L.; Koch, J. Folic acid metabolism. Physiol. Rev. 1967, 47, 83–116. [Google Scholar] [CrossRef]
- Ndaw, S.; Bergaentzlé, M.; Aoudé-Werner, D.; Lahély, S.; Hasselmann, C. Determination of folates in foods by high-performance liquid chromatography with fluorescence detection after precolumn conversion to 5-methyltetrahydrofolates. J. Chromatogr. A 2001, 928, 77–90. [Google Scholar] [CrossRef]
- Hjortmo, S.; Patring, J.; Jastrebova, J.; Andlid, T. Biofortification of folates in white wheat bread by selection of yeast strain and process. Int. J. Food Microbiol. 2008, 127, 32–36. [Google Scholar] [CrossRef]
- Striegel, L.; Chebib, S.; Netzel, M.E.; Rychlik, M. Improved Stable Isotope Dilution Assay for Dietary Folates Using LC-MS/MS and Its Application to Strawberries. Front. Chem. 2018, 6, 6. [Google Scholar] [CrossRef]
- Kopp, M.; Durr, K.; Steigleder, M.; Clavel, T.; Rychlik, M. Development of stable isotope dilution assays for the quantitation of intra- and extracellular folate patterns of Bifidobacterium adolescentis. J. Chromatogr. A 2016, 1469, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Gatto, L.; Lilley, K.S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trivial Name | Abbreviation | R1 | R2 | R3 |
|---|---|---|---|---|
| Pteroylglutamic acid | PteGlu | –N5= | H | –N8= |
| 7,8-dihydrofolate | 7,8-H2folate | H | H | –N8= |
| 5,6,7,8-tetrahydrofolate | 5,6,7,8-H4folate | H | H | H |
| 5-methyl-tetrahydrofolate | 5-CH3-H4folate | CH3 | H | H |
| 5-methyl-dihydrofolate | 5-CH3-H2folate | CH3 | H | –N8= |
| 5-formyl-tetrahydrofolate | 5-CHO-H4folate | CHO | H | H |
| 10-formyl-tetrahydrofolate | 10-CHO-H4folate | H | CHO | H |
| 10-formyl-dihydrofolate | 10-CHO-H2folate | H | CHO | –N8= |
| 10-formyl-pteroic acid | 10-CHO-PteGlu | –N5= | CHO | –N8= |
| 5,10-methenyl-tetrahydrofolate | 5,10-CH+-H4folate | –N5–CH+=N10– | H | |
| 5,10-methylene-tetrahydrofolate | 5,10-CH2-H4folate | –N5–CH2–N10– | H | |
| Procedure | SAXa | SAXb | HLB | C18 |
|---|---|---|---|---|
| pH of sample | 5 | 5 | 2 | 2 |
| Elution solution | 0.1 M NaAc + 5% NaCl, 1% ascorbic acid, 0.1% DTT (dithiothreitol) | 20% ACN + 5% formic acid | MeOH | 20% ACN + 0.1% formic acid |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gmelch, L.; Wirtz, D.; Witting, M.; Weber, N.; Striegel, L.; Schmitt-Kopplin, P.; Rychlik, M. Comprehensive Vitamer Profiling of Folate Mono- and Polyglutamates in Baker’s Yeast (Saccharomyces cerevisiae) as a Function of Different Sample Preparation Procedures. Metabolites 2020, 10, 301. https://doi.org/10.3390/metabo10080301
Gmelch L, Wirtz D, Witting M, Weber N, Striegel L, Schmitt-Kopplin P, Rychlik M. Comprehensive Vitamer Profiling of Folate Mono- and Polyglutamates in Baker’s Yeast (Saccharomyces cerevisiae) as a Function of Different Sample Preparation Procedures. Metabolites. 2020; 10(8):301. https://doi.org/10.3390/metabo10080301
Chicago/Turabian StyleGmelch, Lena, Daniela Wirtz, Michael Witting, Nadine Weber, Lisa Striegel, Philippe Schmitt-Kopplin, and Michael Rychlik. 2020. "Comprehensive Vitamer Profiling of Folate Mono- and Polyglutamates in Baker’s Yeast (Saccharomyces cerevisiae) as a Function of Different Sample Preparation Procedures" Metabolites 10, no. 8: 301. https://doi.org/10.3390/metabo10080301