Metabolic Profiling Reveals Aggravated Non-Alcoholic Steatohepatitis in High-Fat High-Cholesterol Diet-Fed Apolipoprotein E-Deficient Mice Lacking Ron Receptor Signaling


 , , , , and
, , , , and
Abstract
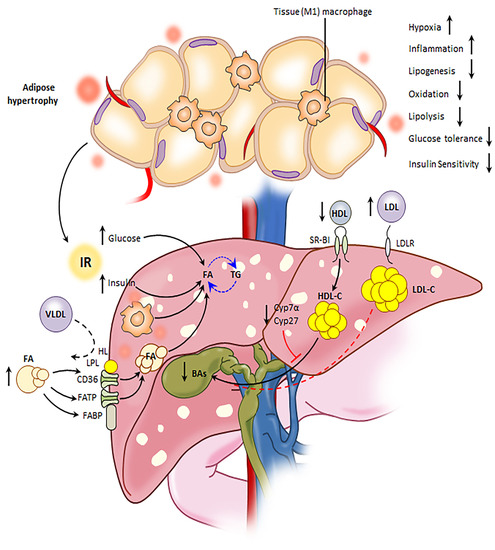
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Loss of Ron Aggravates White Adipose Tissue Metabolic Dysfunction
2.2. Loss of Ron Promotes Enhanced Intrahepatic Fat Storage and Increases Hepatic Fatty Acid Oxidation
2.3. Impaired Ron Receptor Signaling Results in Increased Expression of SREBP-1c and Target Lipogenic Enzymes in Livers of HFHC Diet-Fed Mice
2.4. Ron Receptor Signaling Affects Bile Acid Synthesis and Metabolism in HFHC-Diet-Fed Mice
3. Discussion
4. Materials and Methods
4.1. Animal Model and Diet
4.2. Preparation of Serum, Feces and Tissue
4.3. Glucose Homeostasis Assessment
4.4. Adipocyte Histomorphometry
4.5. Sample Preparation and 1H Nuclear Magnetic Resonance Spectroscopy
4.6. Liver Histopathological Analysis
4.7. RT-PCR Analysis
4.8. GC-MS Analysis of Fatty Acids
4.9. UPLC-MS Analysis of Bile Acids
4.10. Human Liver Gene Expression Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [Green Version]
- Vonghia, L.; Francque, S. Cross talk of the immune system in the adipose tissue and the liver in non-alcoholic steatohepatitis: Pathology and beyond. World J. Hepatol. 2015, 7, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Tessari, P.; Coracina, A.; Cosma, A.; Tiengo, A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 291–302. [Google Scholar] [CrossRef]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and prevention of hepatic steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate immunity and inflammation in nafld/nash. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. Nash is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Chedid, M.F. Nonalcoholic steatohepatitis: The second leading indication for liver transplantation in the USA. Dig. Dis. Sci. 2017, 62, 2621–2622. [Google Scholar] [CrossRef]
- Yu, S.; Allen, J.N.; Dey, A.; Zhang, L.; Balandaram, G.; Kennett, M.J.; Xia, M.; Xiong, N.; Peters, J.M.; Patterson, A.; et al. The ron receptor tyrosine kinase regulates macrophage heterogeneity and plays a protective role in diet-induced obesity, atherosclerosis, and hepatosteatosis. J. Immunol. 2016, 197, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Q.; Chen, Y.Q.; Fisher, J.H.; Wang, M.H. Activation of the ron receptor tyrosine kinase by macrophage-stimulating protein inhibits inducible cyclooxygenase-2 expression in murine macrophages. J. Biol. Chem. 2002, 277, 38104–38110. [Google Scholar] [CrossRef] [Green Version]
- Ray, M.; Yu, S.; Sharda, D.R.; Wilson, C.B.; Liu, Q.; Kaushal, N.; Prabhu, K.S.; Hankey, P.A. Inhibition of tlr4-induced ikappab kinase activity by the ron receptor tyrosine kinase and its ligand, macrophage-stimulating protein. J. Immunol. 2010, 185, 7309–7316. [Google Scholar] [CrossRef]
- Wang, M.H.; Zhou, Y.Q.; Chen, Y.Q. Macrophage-stimulating protein and ron receptor tyrosine kinase: Potential regulators of macrophage inflammatory activities. Scand. J. Immunol. 2002, 56, 545–553. [Google Scholar] [CrossRef]
- Liu, Q.P.; Fruit, K.; Ward, J.; Correll, P.H. Negative regulation of macrophage activation in response to ifn-gamma and lipopolysaccharide by the stk/ron receptor tyrosine kinase. J. Immunol. 1999, 163, 6606–6613. [Google Scholar] [PubMed]
- Chaudhuri, A. Regulation of macrophage polarization by ron receptor tyrosine kinase signaling. Front. Immunol. 2014, 5, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.B.; Ray, M.; Lutz, M.; Sharda, D.; Xu, J.; Hankey, P.A. The ron receptor tyrosine kinase regulates ifn-gamma production and responses in innate immunity. J. Immunol. 2008, 181, 2303–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosello-Trampont, A.-C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Jiao, P.; Nie, Y.; Kim, T.; Jun, D.; van Rooijen, N.; Yang, Z.; Xu, H. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS ONE 2011, 6, e24358. [Google Scholar] [CrossRef]
- Danenberg, H.D.; Fishbein, I.; Gao, J.; Monkkonen, J.; Reich, R.; Gati, I.; Moerman, E.; Golomb, G. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation 2002, 106, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.; Gao, M.; Qu, S.; Liu, D. Intraperitoneal injection of clodronate liposomes eliminates visceral adipose macrophages and blocks high-fat diet-induced weight gain and development of insulin resistance. AAPS J. 2013, 15, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti-Gbaguidi, G.; Staels, B. Macrophage polarization in metabolic disorders: Functions and regulation. Curr. Opin. Lipidol. 2011, 22, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castoldi, A.; De Souza, C.N.; Camara, N.O.; Moraes-Vieira, P.M. The macrophage switch in obesity development. Front. Immunol. 2015, 6, 637. [Google Scholar] [CrossRef] [Green Version]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflugers Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Schierwagen, R.; Maybüchen, L.; Zimmer, S.; Hittatiya, K.; Bäck, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of western diet in apolipoprotein-e-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in nafld/nash. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Xu, X.; Lu, L.; Dong, Q.; Li, X.; Zhang, N.; Xin, Y.; Xuan, S. Research advances in the relationship between nonalcoholic fatty liver disease and atherosclerosis. Lipids Health Dis. 2015, 14, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik-Cichy, K.; Koślińska-Berkan, E.; Piekarska, A. The influence of nafld on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Sharda, D.R.; Yu, S.; Ray, M.; Squadrito, M.L.; De Palma, M.; Wynn, T.A.; Morris, S.M., Jr.; Hankey, P.A. Regulation of macrophage arginase expression and tumor growth by the ron receptor tyrosine kinase. J. Immunol. 2011, 187, 2181–2192. [Google Scholar] [CrossRef]
- Correll, P.H.; Iwama, A.; Tondat, S.; Mayrhofer, G.; Suda, T.; Bernstein, A. Deregulated inflammatory response in mice lacking the stk/ron receptor tyrosine kinase. Genes Funct. 1997, 1, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal models of nonalcoholic fatty liver disease-a starter’s guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bengmark, S.; Qu, S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (nafld). Lipids Health Dis. 2010, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.; Zhang, J.; Quickel, M.D.; Kennett, M.; Patterson, A.D.; Hankey-Giblin, P.A. Ron receptor signaling ameliorates hepatic fibrosis in a diet-induced nonalcoholic steatohepatitis mouse model. J. Proteome Res. 2018, 17, 3268–3280. [Google Scholar] [CrossRef]
- Le Lay, S.; Lefrere, I.; Trautwein, C.; Dugail, I.; Krief, S. Insulin and sterol-regulatory element-binding protein-1c (srebp-1c) regulation of gene expression in 3t3-l1 adipocytes. Identification of ccaat/enhancer-binding protein beta as an srebp-1c target. J. Biol. Chem. 2002, 277, 35625–35634. [Google Scholar] [CrossRef] [Green Version]
- Kolehmainen, M.; Vidal, H.; Alhava, E.; Uusitupa, M.I. Sterol regulatory element binding protein 1c (srebp-1c) expression in human obesity. Obes. Res. 2001, 9, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Carobbio, S.; Hagen, R.M.; Lelliott, C.J.; Slawik, M.; Medina-Gomez, G.; Tan, C.Y.; Sicard, A.; Atherton, H.J.; Barbarroja, N.; Bjursell, M.; et al. Adaptive changes of the insig1/srebp1/scd1 set point help adipose tissue to cope with increased storage demands of obesity. Diabetes 2013, 62, 3697–3708. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, K.J.; Leitner, J.W.; Gurerich, I.; Draznin, B. Insulin regulation of sterol regulatory element-binding protein-1 expression in l-6 muscle cells and 3t3 l1 adipocytes. J. Biol. Chem. 2004, 279, 34380–34387. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Salehi, S.; Cheung, P.; Homko, C.; Song, W.; Loveland-Jones, C.; Jayarajan, S. Comparison of in vivo effects of insulin on srebp-1c activation and insig-1/2 in rat liver and human and rat adipose tissue. Obesity 2013, 21, 1208–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvera, S.; Gealekman, O. Adipose tissue angiogenesis: Impact on obesity and type-2 diabetes. Biochim. Biophys. Acta BBA 2014, 1842, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. 2009, 33, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.-K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.-J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mtorc1-egr1-atgl-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef] [Green Version]
- Jocken, J.W.; Langin, D.; Smit, E.; Saris, W.H.; Valle, C.; Hul, G.B.; Holm, C.; Arner, P.; Blaak, E.E. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J. Clin. Endocrinol. Metab. 2007, 92, 2292–2299. [Google Scholar] [CrossRef] [Green Version]
- Bertola, A.; Bonnafous, S.; Cormont, M.; Anty, R.; Tanti, J.F.; Tran, A.; Le Marchand-Brustel, Y.; Gual, P. Hepatocyte growth factor induces glucose uptake in 3t3-l1 adipocytes through a gab1/phosphatidylinositol 3-kinase/glut4 pathway. J. Biol. Chem. 2007, 282, 10325–10332. [Google Scholar] [CrossRef] [Green Version]
- Muratsu, J.; Iwabayashi, M.; Sanada, F.; Taniyama, Y.; Otsu, R.; Rakugi, H.; Morishita, R. Hepatocyte growth factor prevented high-fat diet-induced obesity and improved insulin resistance in mice. Sci. Rep. 2017, 7, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.G.; Araújo, T.G.; Carvalho, B.M.; Rocha, G.Z.; Santos, A.; Saad, M.J.A. The role of hepatocyte growth factor (hgf) in insulin resistance and diabetes. Front. Endocrinol. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Li, J.; Oligschlaeger, Y.; Jeurissen, M.L.; Houben, T.; Walenbergh, S.M.; Shiri-Sverdlov, R.; Neumann, D. Msp is a negative regulator of inflammation and lipogenesis in ex vivo models of non-alcoholic steatohepatitis. Exp. Mol. Med. 2016, 48, e258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Grunfeld, C. Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J. Clin. Investig. 1987, 80, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Mizuki, E.; Takayuki, M.; Masataka, S.; Hironobu, Y. Tnf-α induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (srebp-1c). Exp. Biol. Med. 2007, 232, 614–621. [Google Scholar]
- Grunfeld, C.; Soued, M.; Adi, S.; Moser, A.H.; Dinarello, C.A.; Feingold, K.R. Evidence for two classes of cytokines that stimulate hepatic lipogenesis: Relationships among tumor necrosis factor, interleukin-1 and interferon-alpha. Endocrinology 1990, 127, 46–54. [Google Scholar] [CrossRef]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver x receptor in insulin-mediated activation of srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Shimano, H.; Amemiya-Kudo, M.; Yahagi, N.; Hasty, A.H.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Identification of liver x receptor-retinoid x receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell. Biol. 2001, 21, 2991–3000. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; So, J.S.; Park, J.G.; Lee, A.H. Transcriptional control of hepatic lipid metabolism by srebp and chrebp. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Azzout-Marniche, D.; Becard, D.; Guichard, C.; Foretz, M.; Ferre, P.; Foufelle, F. Insulin effects on sterol regulatory-element-binding protein-1c (srebp-1c) transcriptional activity in rat hepatocytes. Biochem. J. 2000, 350 Pt 2, 389–393. [Google Scholar] [CrossRef]
- Beltowski, J. Liver x receptors (lxr) as therapeutic targets in dyslipidemia. Cardiovasc. Ther. 2008, 26, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor lxr alpha. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Streicher, R.; Kotzka, J.; Muller-Wieland, D.; Siemeister, G.; Munck, M.; Avci, H.; Krone, W. Srebp-1 mediates activation of the low density lipoprotein receptor promoter by insulin and insulin-like growth factor-i. J. Biol. Chem. 1996, 271, 7128–7133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, D.P.; Knight, B.L.; Soutar, A.K. Regulation of low-density-lipoprotein-receptor mrna by insulin in human hepatoma hep g2 cells. Eur. J. Biochem. 1989, 181, 727–731. [Google Scholar] [CrossRef]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.H.; Gavrilova, O.; Ward, J.M.; Brewer, B., Jr.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of ppargamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Investig. 2003, 111, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter cd36 is a common target of lxr, pxr, and ppargamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef]
- McArthur, M.J.; Atshaves, B.P.; Frolov, A.; Foxworth, W.D.; Kier, A.B.; Schroeder, F. Cellular uptake and intracellular trafficking of long chain fatty acids. J. Lipid Res. 1999, 40, 1371–1383. [Google Scholar]
- Motojima, K.; Passilly, P.; Peters, J.M.; Gonzalez, F.J.; Latruffe, N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J. Biol. Chem. 1998, 273, 16710–16714. [Google Scholar] [CrossRef] [Green Version]
- Börchers, T.; Spener, F. Fatty acid binding proteins. Curr. Top. Membr. 1994, 40, 261–294. [Google Scholar]
- Shimada, M.; Shimano, H.; Gotoda, T.; Yamamoto, K.; Kawamura, M.; Inaba, T.; Yazaki, Y.; Yamada, N. Overexpression of human lipoprotein lipase in transgenic mice. Resistance to diet-induced hypertriglyceridemia and hypercholesterolemia. J. Biol. Chem. 1993, 268, 17924–17929. [Google Scholar]
- Excoffon, K.J.; Liu, G.; Miao, L.; Wilson, J.E.; McManus, B.M.; Semenkovich, C.F.; Coleman, T.; Benoit, P.; Duverger, N.; Branellec, D.; et al. Correction of hypertriglyceridemia and impaired fat tolerance in lipoprotein lipase-deficient mice by adenovirus-mediated expression of human lipoprotein lipase. Arter. Thromb. Vasc. Biol. 1997, 17, 2532–2539. [Google Scholar] [CrossRef] [PubMed]
- Dichek, H.L.; Brecht, W.; Fan, J.; Ji, Z.S.; McCormick, S.P.; Akeefe, H.; Conzo, L.; Sanan, D.A.; Weisgraber, K.H.; Young, S.G.; et al. Overexpression of hepatic lipase in transgenic mice decreases apolipoprotein b-containing and high density lipoproteins. Evidence that hepatic lipase acts as a ligand for lipoprotein uptake. J. Biol. Chem. 1998, 273, 1896–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, T.; Czimmerer, Z.; Nagy, L. Ppars are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta BBA 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Marion-Letellier, R.; Savoye, G.; Ghosh, S. Fatty acids, eicosanoids and ppar gamma. Eur. J. Pharmacol. 2016, 785, 44–49. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Mardones, P.; Quinones, V.; Amigo, L.; Moreno, M.; Miquel, J.F.; Schwarz, M.; Miettinen, H.E.; Trigatti, B.; Krieger, M.; VanPatten, S.; et al. Hepatic cholesterol and bile acid metabolism and intestinal cholesterol absorption in scavenger receptor class b type i-deficient mice. J. Lipid Res. 2001, 42, 170–180. [Google Scholar]
- Wiersma, H.; Gatti, A.; Nijstad, N.; Kuipers, F.; Tietge, U.J.F. Hepatic sr-bi, not endothelial lipase, expression determines biliary cholesterol secretion in mice. J. Lipid Res. 2009, 50, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, H.; Gatti, A.; Nijstad, N.; Oude Elferink, R.P.; Kuipers, F.; Tietge, U.J. Scavenger receptor class b type i mediates biliary cholesterol secretion independent of atp-binding cassette transporter g5/g8 in mice. Hepatology 2009, 50, 1263–1272. [Google Scholar] [CrossRef]
- Herscovitz, H.; Ronen, I.; Bilu, S.; Tietz, A. Bile acid synthesis from hdl cholesterol and cholesterol ester by cultured chick embryo hepatocytes. Biochim. Biophys. Acta BBA 1986, 878, 426–434. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, N.; Ramakrishnan, R.; Sehayek, E.; Huszar, D.; Breslow, J.L.; Tall, A.R. Hepatic scavenger receptor bi promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J. Biol. Chem. 1999, 274, 33398–33402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portman, O.W.; Alexander, M.; O’Malley, J.P. Metabolism of free and esterified cholesterol and apolipoproteins of plasma low and high density lipoproteins. Biochim. Biophys. Acta BBA 1980, 619, 545–558. [Google Scholar] [CrossRef]
- Miller, L.K.; Tiell, M.L.; Paul, I.; Spaet, T.H.; Rosenfeld, R.S. Side-chain oxidation of lipoprotein-bound [24,25-3h]cholesterol in the rat: Comparison of hdl and ldl and implications for bile acid synthesis. J. Lipid Res. 1982, 23, 335–344. [Google Scholar] [PubMed]
- Schwartz, C.C.; Vlahcevic, Z.R.; Halloran, L.G.; Swell, L. An in vivo evaluation in man of the transfer of esterified cholesterol between lipoproteins and into the liver and bile. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1981, 663, 143–162. [Google Scholar] [CrossRef]
- Shin, D.J.; Osborne, T.F. Fgf15/fgfr4 integrates growth factor signaling with hepatic bile acid metabolism and insulin action. J. Biol. Chem. 2009, 284, 11110–11120. [Google Scholar] [CrossRef] [Green Version]
- Claudel, T.; Trauner, M. Bile acids and their receptors. In Signaling Pathways in Liver Diseases; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 385–399. [Google Scholar]
- Twisk, J.; Hoekman, M.F.M.; Lehmann, E.M.; Meijer, P.; Mager, W.H.; Princen, H.M.G. Insulin suppresses bile acid synthesis in cultured rat hepatocytes by down-regulation of cholesterol 7α-hydroxylase and sterol 27-hydroxylase gene transcription. Hepatology 1995, 21, 501–510. [Google Scholar] [CrossRef]
- Wang, D.P.; Stroup, D.; Marrapodi, M.; Crestani, M.; Galli, G.; Chiang, J.Y. Transcriptional regulation of the human cholesterol 7 alpha-hydroxylase gene (cyp7a) in hepg2 cells. J. Lipid Res. 1996, 37, 1831–1841. [Google Scholar]
- Reid, D.T.; Reyes, J.L.; McDonald, B.A.; Vo, T.; Reimer, R.A.; Eksteen, B. Kupffer cells undergo fundamental changes during the development of experimental nash and are critical in initiating liver damage and inflammation. PLoS ONE 2016, 11, e0159524. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.-Y.; Kim, D.-H.; Chun, K.-H. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab. Anim. Res. 2018, 34, 133–139. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through ccr2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [Green Version]
- Alharthi, J.; Latchoumanin, O.; George, J.; Eslam, M. Macrophages in metabolic associated fatty liver disease. World J. Gastroenterol. 2020, 26, 1861–1878. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Kwon, M.-J.; Choi, A.M.K.; Kim, H.-P.; Nakahira, K.; Hwang, D.H. Fatty acids modulate toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J. Boil. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [Green Version]
- Rosso, C.; Kazankov, K.; Younes, R.; Esmaili, S.; Marietti, M.; Sacco, M.; Carli, F.; Gaggini, M.; Salomone, F.; Møller, H.J.; et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J. Hepatol. 2019, 71, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. Tlr4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [PubMed] [Green Version]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate tlr-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezerra, J.A.; Laney, D.W.; Degen, S.J.F. Increased expression of mrna for hepatocyte growth factor-like protein during liver regeneration and inflammation. Biochem. Biophys. Res. Commun. 1994, 203, 666–673. [Google Scholar] [CrossRef]
- Locaputo, S.; Carrick, T.L.; Bezerra, J.A. Zonal regulation of gene expression during liver regeneration of urokinase transgenic mice. Hepatology 1999, 29, 1106–1113. [Google Scholar] [CrossRef]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (hgf) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-D.; Kim, S.-S.; Cha, H.-Y.; Jang, S.-H.; Chang, D.-Y.; Kim, W.; Suh-Kim, H.; Lee, J.-H. Therapeutic effect of hepatocyte growth factor-secreting mesenchymal stem cells in a rat model of liver fibrosis. Exp. Mol. Med. 2014, 46, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, T.; Kaneda, Y.; Tsutsui, H.; Nakanishi, K.; Sawa, Y.; Morishita, R.; Matsumoto, K.; Nakamura, T.; Takahashi, H.; Okamoto, E.; et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat. Med. 1999, 5, 226–230. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat. Clin. Pract. Gastroenterol. Hepatol. 2004, 1, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Drescher, H.K.; Schumacher, F.; Schenker, T.; Baues, M.; Lammers, T.; Hieronymus, T.; Trautwein, C.; Streetz, K.L.; Kroy, D.C. C-met signaling protects from nonalcoholic steatohepatitis- (nash-) induced fibrosis in different liver cell types. Oxid. Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Dou, Z.; Liu, J.; Chai, B.; Li, Y.; An, X.; Chu, P.; Zhang, X. Therapeutic effect of hgf on nash mice through hgf/c-met and jak2-stat3 signalling pathway. Ann. Hepatol. 2018, 17, 501–510. [Google Scholar] [CrossRef]
- Kizu, T.; Yoshida, Y.; Furuta, K.; Ogura, S.; Egawa, M.; Chatani, N.; Hamano, M.; Takemura, T.; Ezaki, H.; Kamada, Y.; et al. Loss of gab1 adaptor protein in hepatocytes aggravates experimental liver fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G613–G624. [Google Scholar] [CrossRef] [Green Version]
- Furuta, K.; Yoshida, Y.; Ogura, S.; Kurahashi, T.; Kizu, T.; Maeda, S.; Egawa, M.; Chatani, N.; Nishida, K.; Nakaoka, Y.; et al. Gab1 adaptor protein acts as a gatekeeper to balance hepatocyte death and proliferation during acetaminophen-induced liver injury in mice. Hepatology 2016, 63, 1340–1355. [Google Scholar] [CrossRef]
- Krawczyk, M.; Zimmermann, S.; Hess, G.; Holz, R.; Dauer, M.; Raedle, J.; Lammert, F.; Grunhage, F. Panel of three novel serum markers predicts liver stiffness and fibrosis stages in patients with chronic liver disease. PLoS ONE 2017, 12, e0173506. [Google Scholar]
- Cuneo, K.C.; Devasia, T.; Sun, Y.; Schipper, M.J.; Karnak, D.; Davis, M.A.; Owen, D.; Feng, M.; El Naqa, I.; Bazzi, L.; et al. Serum levels of hepatocyte growth factor and cd40 ligand predict radiation-induced liver injury. Transl. Oncol. 2019, 12, 889–894. [Google Scholar] [CrossRef]
- Shiota, G.; Okano, J.-I.; Kawasaki, H.; Kawamoto, T.; Nakamura, T. Serum hepatocyte growth factor levels in liver diseases: Clinical implications. Hepatology 1995, 21, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Masuhara, M.; Yasunaga, M.; Tanigawa, K.; Tamura, F.; Yamashita, S.; Sakaida, I.; Okita, K. Expression of hepatocyte growth factor, transforming growth factor α, and transforming growth factor β 1 messenger rna in various human liver diseases and correlation with hepatocyte proliferation. Hepatology 1996, 24, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, J.A. Therapeutic targets in liver fibrosis. Am. J. Physiol. Liver Physiol. 2011, 300, G709–G715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xie, C.; Nichols, R.G.; Chan, S.H.; Jiang, C.; Hao, R.; Smith, P.B.; Cai, J.; Simons, M.N.; Hatzakis, E.; et al. Farnesoid x receptor signaling shapes the gut microbiota and controls hepatic lipid metabolism. mSystems 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarafian, M.H.; Lewis, M.R.; Pechlivanis, A.; Ralphs, S.; McPhail, M.J.; Patel, V.C.; Dumas, M.E.; Holmes, E.; Nicholson, J.K. Bile acid profiling and quantification in biofluids using ultra-performance liquid chromatography tandem mass spectrometry. Anal. Chem. 2015, 87, 9662–9670. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-beta hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, J.N.; Dey, A.; Cai, J.; Zhang, J.; Tian, Y.; Kennett, M.; Ma, Y.; Liang, T.J.; Patterson, A.D.; Hankey-Giblin, P.A. Metabolic Profiling Reveals Aggravated Non-Alcoholic Steatohepatitis in High-Fat High-Cholesterol Diet-Fed Apolipoprotein E-Deficient Mice Lacking Ron Receptor Signaling. Metabolites 2020, 10, 326. https://doi.org/10.3390/metabo10080326
Allen JN, Dey A, Cai J, Zhang J, Tian Y, Kennett M, Ma Y, Liang TJ, Patterson AD, Hankey-Giblin PA. Metabolic Profiling Reveals Aggravated Non-Alcoholic Steatohepatitis in High-Fat High-Cholesterol Diet-Fed Apolipoprotein E-Deficient Mice Lacking Ron Receptor Signaling. Metabolites. 2020; 10(8):326. https://doi.org/10.3390/metabo10080326
Chicago/Turabian StyleAllen, Joselyn N., Adwitia Dey, Jingwei Cai, Jingtao Zhang, Yuan Tian, Mary Kennett, Yanling Ma, T. Jake Liang, Andrew D. Patterson, and Pamela A. Hankey-Giblin. 2020. "Metabolic Profiling Reveals Aggravated Non-Alcoholic Steatohepatitis in High-Fat High-Cholesterol Diet-Fed Apolipoprotein E-Deficient Mice Lacking Ron Receptor Signaling" Metabolites 10, no. 8: 326. https://doi.org/10.3390/metabo10080326