Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches

Abstract
:1. Introduction
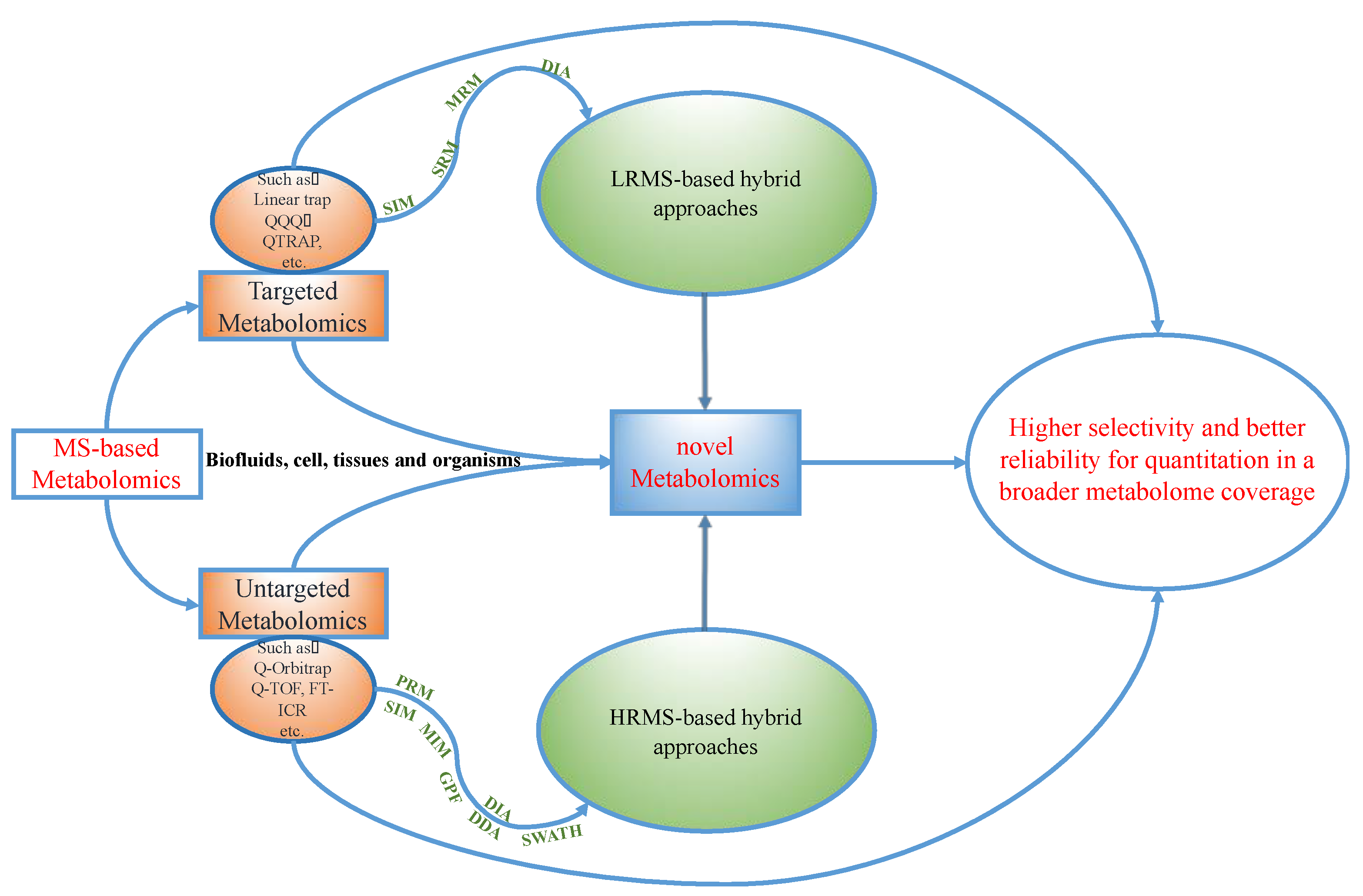
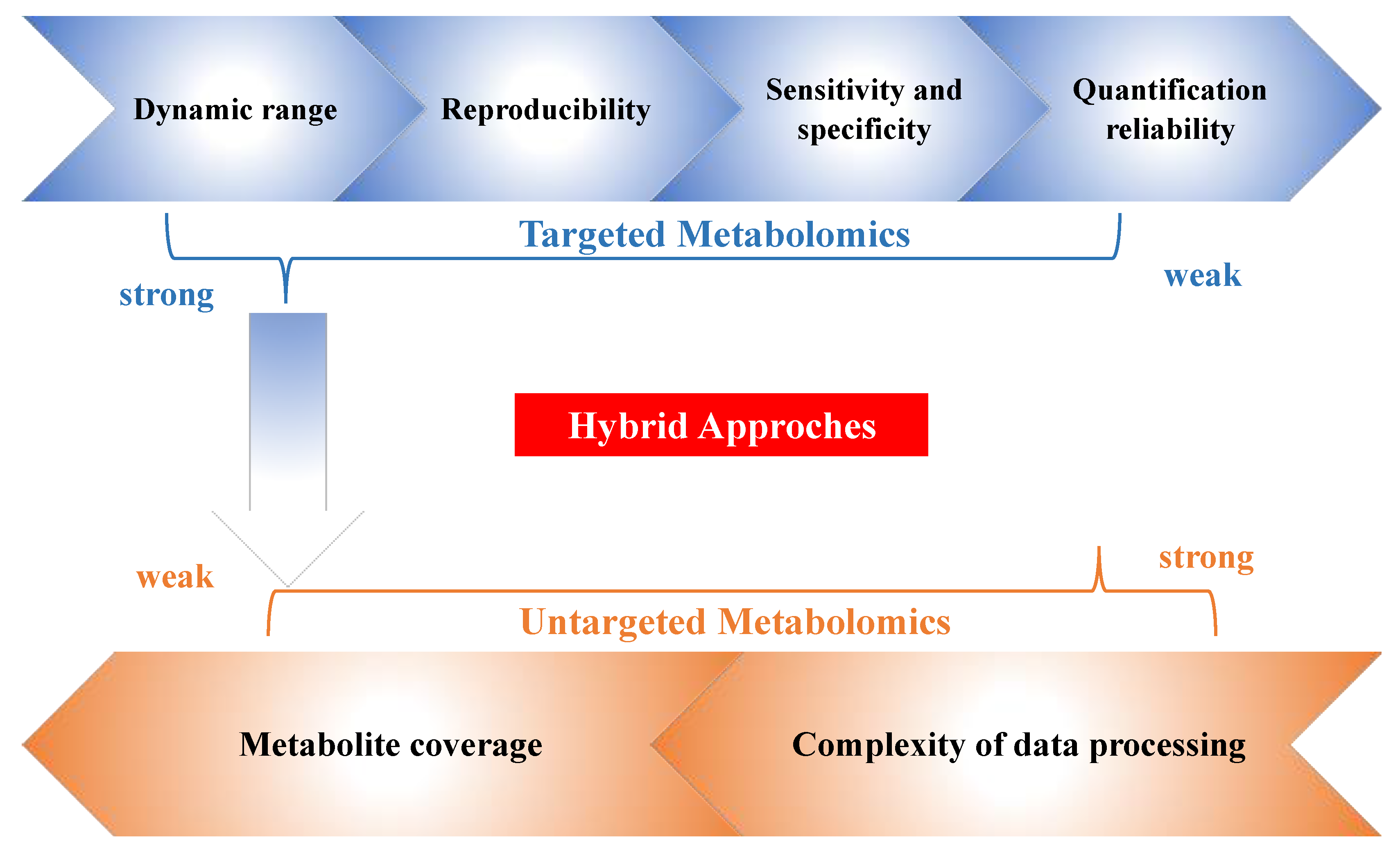
2. Hybrid Approaches and the Novel Workflow in Metabolomics
2.1. The Collection of Ion Pairs List in Hybrid Approaches
2.2. The Choices of Metabolomics Platforms in Hybrid Analyses
2.2.1. The Single Usage of LRMS with Hybrid Approaches
2.2.2. The Single Usage of HRMS with Hybrid Approaches
2.2.3. The Integration of LRMS and HRMS with Hybrid Approaches
2.3. Data Handling in Hybrid Metabolomics Approches
3. Representative Applications of Hybrid Approaches in Metabolomics
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMDIS | automated mass spectral deconvolution and identification system |
| BC | bladder cancer |
| cv | coefficient of variation |
| DDA | data-dependent acquisition |
| tDDA | target-direct data-dependent acquisition |
| tsDDA | time staggered-direct data-dependent acquisition |
| DITQM | data independent targeted quantitative metabolomics |
| EIC | extracted ions current |
| ESI | electrospray ionization quadrupole |
| GC | gas chromatography |
| GC-MS | gas chromatography coupled with mass spectrometry |
| GOT | globally optimized targeted method |
| dGOT | database-assisted globally optimized targeted method |
| ts/msGOT | time staggered/mass staggered globally optimized targeted method |
| GPF | gas-phase fractionation method |
| HCC | hepatocellular carcinoma |
| HMDB | human metabolome database |
| HPLC | high-performance liquid chromatography |
| UHPLC | Ulrta-high performance liquid chromatography |
| HPLC-MS | high-performance liquid chromatography coupled with mass spectrometry |
| HRMS | high resolution mass spectrometers |
| IDA | information dependent acquisition |
| LRMS | low resolution mass spectrometers |
| MIM | multiple ion monitoring |
| tsMIM | time staggered-multiple ion monitoring |
| MRM | multiple reaction monitoring |
| MS | mass spectrometry |
| NMR | nuclear magnetic resonance |
| Q-Orbitrap | quadrupole-orbitrap |
| Q-TOF | quadrupole time-of-flight |
| QC | quality control |
| QQQ MS | triple quadrupole mass spectrometry |
| QTrap | quadrupole trap |
| RSD | Relative Standard Deviation |
| RT | retention time |
| RTL | the retention time locking |
| SESI | secondary electrospray ionization |
| SIM | selected ion monitoring |
| SRM | selected reaction monitoring |
| SWATH | sequential windowed acquisition of all theoretical fragment ion |
| TIC | total ion current |
| TOF | time of flight |
References
- Nicholson, J.K.; Wilson, I.D. Understanding’global’systems biology: Metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003, 2, 668. [Google Scholar] [CrossRef] [PubMed]
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 1961, 3, 208–210. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Recent and potential developments of biofluid analyses in metabolomics. J. Proteom. 2012, 75, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Z.; Lu, X.; Yang, X.; Yin, P.; Kong, H.; Yu, Y.; Xu, G. Comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry for metabonomics: Biomarker discovery for diabetes mellitus. Anal. Chim. Acta 2009, 633, 257–262. [Google Scholar] [CrossRef]
- Carrola, J.; Rocha, C.U.M.; Barros, A.N.S.; Gil, A.M.; Goodfellow, B.J.; Carreira, I.M.; Bernardo, J.; Gomes, A.; Sousa, V.; Carvalho, L. Metabolic signatures of lung cancer in biofluids: NMR-based metabonomics of urine. J. Proteome Res. 2011, 10, 221–230. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A global biochemical approach to drug response and disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.-P.; Collino, S.; Rezzi, S.; Kochhar, S. Metabolomic applications to decipher gut microbial metabolic influence in health and disease. Front. Physiol. 2012, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.G.; Reily, M.D.; Sigler, R.E.; Wells, D.F.; Paterson, D.A.; Braden, T.K. Metabonomics: Evaluation of nuclear magnetic resonance (NMR) and pattern recognition technology for rapid in vivo screening of liver and kidney toxicants. Toxicol. Sci. 2000, 57, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. [Google Scholar] [CrossRef]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar] [CrossRef]
- Orians, K.J.; Boyle, E.A. Determination of picomolar concentrations of titanium, gallium and indium in sea water by inductively coupled plasma mass spectrometry following an 8-hydroxyquinoline chelating resin preconcentration. Anal. Chim. Acta 1993, 282, 63–74. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Modern analytical techniques in metabolomics analysis. Analyst 2012, 137, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Bennett, B.D.; Rabinowitz, J.D. Analytical strategies for LC–MS-based targeted metabolomics. J. Chromatogr. B 2008, 871, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, M.G.; Nix, C.; Nys, G.; Fillet, M.J.T. Targeted metabolomics of whole blood using volumetric absorptive microsampling. Talanta 2019, 197, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kremer, D.M.; Sajjakulnukit, P.; Zhang, L.; Lyssiotis, C.A. A large-scale analysis of targeted metabolomics data from heterogeneous biological samples provides insights into metabolite dynamics. Metabolomics 2019, 15, 103. [Google Scholar] [CrossRef] [Green Version]
- Breier, M.; Wahl, S.; Prehn, C.; Fugmann, M.; Ferrari, U.; Weise, M.; Banning, F.; Seissler, J.; Grallert, H.; Adamski, J.; et al. Targeted Metabolomics Identifies Reliable and Stable Metabolites in Human Serum and Plasma Samples. PLoS ONE 2014, 9, e89728. [Google Scholar] [CrossRef]
- Warth, B.; Parich, A.; Bueschl, C.; Schoefbeck, D.; Neumann, N.K.N.; Kluger, B.; Schuster, K.; Krska, R.; Adam, G.; Lemmens, M. GC–MS based targeted metabolic profiling identifies changes in the wheat metabolome following deoxynivalenol treatment. Metabolomics 2015, 11, 722–738. [Google Scholar] [CrossRef] [Green Version]
- Mazzini, F.N.; Cook, F.; Gounarides, J.; Marciano, S.; Haddad, L.; Tamaroff, A.J.; Casciato, P.; Narvaez, A.; Mascardi, M.F.; Anders, M.J.M. Plasma and stool metabolomic biomarkers of non-alcoholic fatty liver disease in Argentina. medRxiv 2020. [Google Scholar] [CrossRef]
- Kuhring, M.; Eisenberger, A.; Schmidt, V.; Kränkel, N.; Leistner, D.M.; Kirwan, J.; Beule, D.J.B. Concepts and software package for efficient quality control in targeted metabolomics studies–MeTaQuaC. Anal. Chem. 2020, 92, 10241–10245. [Google Scholar] [CrossRef]
- Rocchetti, G.; Giuberti, G.; Busconi, M.; Marocco, A.; Trevisan, M.; Lucini, L. Pigmented sorghum polyphenols as potential inhibitors of starch digestibility: An in vitro study combining starch digestion and untargeted metabolomics. Food Chem. 2020, 312, 126077. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X.; Xu, L.; Ran, X.; Li, X.; Chen, L.; Zhao, X.; Deng, H.; Liu, X. Establishment of local searching methods for orbitrap-based high throughput metabolomics analysis. Talanta 2016, 156, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Erban, A.; Weber, R.J.; Creek, D.J.; Brown, M.; Breitling, R.; Hankemeier, T.; Goodacre, R.; Neumann, S.; Kopka, J. Mass appeal: Metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9, 44–66. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.F.; Zhou, B.; Ressom, H.W. Metabolite identification and quantitation in LC-MS/MS-based metabolomics. Trac Trends Anal. Chem. 2012, 32, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Zhang, P.; Zhu, J.; Raftery, D. Globally optimized targeted mass spectrometry: Reliable metabolomics analysis with broad coverage. Anal. Chem. 2015, 87, 12355–12362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ruan, Q.; Li, Y.; Ye, G.; Lu, X.; Lin, X.; Xu, G. A novel approach to transforming a non-targeted metabolic profiling method to a pseudo-targeted method using the retention time locking gas chromatography/mass spectrometry-selected ions monitoring. J. Chromatogr. A 2012, 1255, 228–236. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Yue, X.; He, J.; Zhang, R.; Xu, J.; Zhou, Z.; Wang, Z.; Zhang, R.; Abliz, Z. Development of simultaneous targeted metabolite quantification and untargeted metabolomics strategy using dual-column liquid chromatography coupled with tandem mass spectrometry. Anal. Chim. Acta 2018, 1037, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, F.; Li, P.; He, C.; Wang, R.; Su, H.; Wan, J.-B. An improved pseudotargeted metabolomics approach using multiple ion monitoring with time-staggered ion lists based on ultra-high performance liquid chromatography/quadrupole time-of-flight mass spectrometry. Anal. Chim. Acta 2016, 927, 82–88. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, C.; Zeng, Z.; Luo, P.; Zhao, Y.; Zhao, J.; Li, L.; Lu, X.; Xu, G. Sample-directed pseudotargeted method for the metabolic profiling analysis of rice seeds based on liquid chromatography with mass spectrometry. J. Sep. Sci. 2016, 39, 247–255. [Google Scholar] [CrossRef]
- Li, H.; Xu, M.; Zhu, J. Headspace gas monitoring of gut microbiota using targeted and globally optimized targeted secondary electrospray ionization mass spectrometry. Anal. Chem. 2018, 91, 854–863. [Google Scholar] [CrossRef]
- Theodoridis, G.; Gika, H.G.; Wilson, I.D. LC-MS-based methodology for global metabolite profiling in metabonomics/metabolomics. Trac Trends Anal. Chem. 2008, 27, 251–260. [Google Scholar] [CrossRef]
- Luo, P.; Dai, W.; Yin, P.; Zeng, Z.; Kong, H.; Zhou, L.; Wang, X.; Chen, S.; Lu, X.; Xu, G. Multiple reaction monitoring-ion pair finder: A systematic approach to transform nontargeted mode to pseudotargeted mode for metabolomics study based on liquid chromatography–mass spectrometry. Anal. Chem. 2015, 87, 5050–5055. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Akiyama, K.; Sakata, A.; Kuwahara, A.; Otsuki, H.; Sakurai, T.; Saito, K.; Hirai, M.Y. Widely targeted metabolomics based on large-scale MS/MS data for elucidating metabolite accumulation patterns in plants. Plant Cell Physiol. 2009, 50, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Buescher, J.M.; Moco, S.; Sauer, U.; Zamboni, N. Ultrahigh performance liquid chromatography− tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Anal. Chem. 2010, 82, 4403–4412. [Google Scholar] [CrossRef]
- Penner, N.; Ramanathan, R.; Zgoda-Pols, J.; Chowdhury, S. Quantitative determination of hippuric and benzoic acids in urine by LC–MS/MS using surrogate standards. J. Pharm. Biomed. Anal. 2010, 52, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Ståhlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Borén, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2009, 877, 2664–2672. [Google Scholar] [CrossRef]
- Li, Y.; Pang, T.; Li, Y.; Ye, G.; Lu, X.; Xu, G. Chemical properties investigation of commercial cigarettes by a “pseudo” targeted method using GC-MS-selected ions monitoring. J. Sep. Sci. 2013, 36, 1545–1552. [Google Scholar] [CrossRef]
- Chen, S.; Kong, H.; Lu, X.; Li, Y.; Yin, P.; Zeng, Z.; Xu, G. Pseudotargeted metabolomics method and its application in serum biomarker discovery for hepatocellular carcinoma based on ultra high-performance liquid chromatography/triple quadrupole mass spectrometry. Anal. Chem. 2013, 85, 8326–8333. [Google Scholar] [CrossRef]
- Calderón-Santiago, M.N.; Priego-Capote, F.; Luque de Castro, M.A.D. Enhanced detection and identification in metabolomics by use of LC–MS/MS untargeted analysis in combination with gas-phase fractionation. Anal. Chem. 2014, 86, 7558–7565. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Yang, W.; Bi, N.; Xu, J.; He, J.; Zhang, R.; Wang, L.; Abliz, Z. Development of a Data-Independent Targeted Metabolomics Method for Relative Quantification Using Liquid Chromatography Coupled with Tandem Mass Spectrometry. Anal. Chem. 2017, 89, 6954–6962. [Google Scholar] [CrossRef]
- Shao, Y.; Zhu, B.; Zheng, R.; Zhao, X.; Yin, P.; Lu, X.; Jiao, B.; Xu, G.; Yao, Z. Development of urinary pseudotargeted LC-MS-based metabolomics method and its application in hepatocellular carcinoma biomarker discovery. J. Proteome Res. 2015, 14, 906–916. [Google Scholar] [CrossRef]
- Liu, P.; Qi, C.-B.; Zhu, Q.-F.; Yuan, B.-F.; Feng, Y.-Q. Determination of thiol metabolites in human urine by stable isotope labeling in combination with pseudo-targeted mass spectrometry analysis. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Feng, R.; Wang, R.; Yang, F.; Li, P.; Wan, J.-B. Enhanced MS/MS coverage for metabolite identification in LC-MS-based untargeted metabolomics by target-directed data dependent acquisition with time-staggered precursor ion list. Anal. Chim. Acta 2017, 992, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, R.; He, C.; Su, H.; Ma, H.; Wan, J.-B. An integrated strategy to improve data acquisition and metabolite identification by time-staggered ion lists in UHPLC/Q-TOF MS-based metabolomics. J. Pharm. Biomed. Anal. 2018, 157, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Q.; Hu, C.; Yu, D.; Wang, L.; Zhou, Y.; Zhao, X.; Li, Q.; Hou, X.; Xu, G. Development of a High Coverage Pseudotargeted Lipidomics Method Based on Ultra-High Performance Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2018, 90, 7608–7616. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xu, M.; Zhu, J. Development and application of time staggered/mass staggered-globally optimized targeted mass spectrometry. J. Chromatogr. B 2019, 1120, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.G.; Hendrickson, C.L. High-resolution mass spectrometers. Annu. Rev. Anal. Chem. 2008, 1, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Han, Y.; Wang, X. Metabolomics in diagnosis and biomarker discovery of colorectal cancer. Cancer Lett. 2014, 345, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Han, L.; Luo, M.; Bian, B.; Guan, M.; Yang, H.; Han, C.; Li, N.; Li, T.; Li, S. Multi-component identification and target cell-based screening of potential bioactive compounds in toad venom by UPLC coupled with high-resolution LTQ-Orbitrap MS and high-sensitivity Qtrap MS. Anal. Bioanal. Chem. 2018, 410, 4419–4435. [Google Scholar] [CrossRef]
- Hernandez, F.; Sancho, J.; Pozo, O. Critical review of the application of liquid chromatography/mass spectrometry to the determination of pesticide residues in biological samples. Anal. Bioanal. Chem. 2005, 382, 934–946. [Google Scholar] [CrossRef]
- Perez, E.R.; Knapp, J.A.; Horn, C.K.; Stillman, S.L.; Evans, J.E.; Arfsten, D.P. Comparison of LC–MS-MS and GC–MS analysis of benzodiazepine compounds included in the drug demand reduction urinalysis program. J. Anal. Toxicol. 2016, 40, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Wang, S.; Jasbi, P.; Turner, C.; Hrovat, J.; Wei, Y.; Liu, J.; Gu, H. Database-Assisted Globally Optimized Targeted Mass Spectrometry (dGOT-MS): Broad and Reliable Metabolomics Analysis with Enhanced Identification. Anal. Chem. 2019, 91, 13737–13745. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, R.; Ma, C.; Zhou, L.; Liu, X.; Yin, P.; Zhang, Z.; Sun, Y.; Xu, C.; Lu, X. Discovery and validation of potential urinary biomarkers for bladder cancer diagnosis using a pseudotargeted GC-MS metabolomics method. Oncotarget 2017, 8, 20719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, J.; Zhang, R.; He, J.; Chen, Y.; Bi, N.; Song, Y.; Wang, L.; Zhan, Q.; Abliz, Z. Development of a metabolic pathway-based pseudo-targeted metabolomics method using liquid chromatography coupled with mass spectrometry. Talanta 2019, 192, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Yin, P.; Hua, R.; Tan, Y.; Li, Z.; Qiu, G.; Yin, Z.; Xie, X.; Wang, X.; Chen, W. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology 2018, 67, 662–675. [Google Scholar] [CrossRef]
- Wang, L.; Su, B.; Zeng, Z.; Li, C.; Zhao, X.; Lv, W.; Xuan, Q.; Ouyang, Y.; Zhou, L.; Yin, P. Ion-pair selection method for pseudotargeted metabolomics based on SWATH MS acquisition and its application in differential metabolite discovery of type 2 diabetes. Anal. Chem. 2018, 90, 11401–11408. [Google Scholar] [CrossRef] [Green Version]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E. METLIN: A technology platform for identifying knowns and unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: The roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef]
- Dhiman, N.; Hall, L.; Wohlfiel, S.L.; Buckwalter, S.P.; Wengenack, N.L. Performance and cost analysis of matrix-assisted laser desorption ionization–time of flight mass spectrometry for routine identification of yeast. J. Clin. Microbiol. 2011, 49, 1614–1616. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, C.; Simó, C.; García-Cañas, V.; Gómez-Martínez, Á.; Ferragut, J.A.; Cifuentes, A. CE/LC-MS multiplatform for broad metabolomic analysis of dietary polyphenols effect on colon cancer cells proliferation. Electrophoresis 2012, 33, 2328–2336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.-Y.; Liu, J.; Cheng, X.-L.; Bai, X.; Lin, R.-C. Urinary metabonomics study on biochemical changes in an experimental model of chronic renal failure by adenine based on UPLC Q-TOF/MS. Clin. Chim. Acta 2012, 413, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Foster, L.J.; de Hoog, C.L.; Mann, M. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. USA 2003, 100, 5813–5818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Jing, T.; Zhao, M.; Jin, J.; Xu, M.; Chen, Y.; Zhang, S.; Wan, X.; Schwab, W.; Song, C.K. Untargeted metabolomics coupled with chemometrics analysis reveals potential non-volatile markers during oolong tea shaking. Food Res. Int. 2019, 123, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, Y.; Zhu, Z.-J. Advancing untargeted metabolomics using data-independent acquisition mass spectrometry technology. Anal. Bioanal. Chem. 2019, 411, 4349–4357. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Smelter, A.; Moseley, H.N.B. A Python library for FAIRer access and deposition to the Metabolomics Workbench Data Repository. Metabolomics 2018, 14, 64. [Google Scholar] [CrossRef] [Green Version]
- Warrack, B.M.; Hnatyshyn, S.; Ott, K.-H.; Reily, M.D.; Sanders, M.; Zhang, H.; Drexler, D.M. Normalization strategies for metabonomic analysis of urine samples. J. Chromatogr. B 2009, 877, 547–552. [Google Scholar] [CrossRef]
- Bijlsma, S.; Bobeldijk, I.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; Van Ommen, B.; Smilde, A.K. Large-scale human metabolomics studies: A strategy for data (pre-) processing and validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate analysis in metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar]
- Saccenti, E.; Hoefsloot, H.C.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Yeung, P.J.A.N.Z.S.W. SPSS Survival Manual. A Step by Step Guide to Data Analysis Using IBM SPSS; Routledge: Abingdon-on-Thames, UK, 2014; Volume 26, p. 92. [Google Scholar]
- Prism, G. Graphpad Software; Prism, GraphPad: San Diegocausa, CA, USA, 1994. [Google Scholar]
- Umetri, A. SIMCA-P for Windows, Graphical Software for Multivariate Process Modeling; Umeasweden: Umea, Sweden, 1996. [Google Scholar]
- Rebholz, C.M.; Rhee, E.P. Application of Metabolomics to Renal and Cardiometabolic Diseases. In Computational Methods and Data Analysis for Metabolomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 401–417. [Google Scholar]
- Niedzwiecki, M.M.; Walker, D.I.; Howell, J.C.; Watts, K.D.; Jones, D.P.; Miller, G.W.; Hu, W.T. High-resolution metabolomic profiling of Alzheimer’s disease in plasma. Ann. Clin. Transl. Neurol. 2020, 7, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, R.F.; Rhee, E.P. Proteomics and metabolomics in kidney disease, including insights into etiology, treatment, and prevention. Clin. J. Am. Soc. Nephrol. 2020, 15, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Gómez, D.; Martínez-Lozano Sinues, P.; Barrios-Collado, C.S.; Vidal-de-Miguel, G.; Gaugg, M.; Zenobi, R. Identification of 2-alkenals, 4-hydroxy-2-alkenals, and 4-hydroxy-2, 6-alkadienals in exhaled breath condensate by UHPLC-HRMS and in breath by real-time HRMS. Anal. Chem. 2015, 87, 3087–3093. [Google Scholar] [CrossRef]
- Zhu, J.; Jiménez-Díaz, J.; Bean, H.D.; Daphtary, N.A.; Aliyeva, M.I.; Lundblad, L.K.; Hill, J.E. Robust detection of P. aeruginosa and S. aureus acute lung infections by secondary electrospray ionization-mass spectrometry (SESI-MS) breathprinting: From initial infection to clearance. J. Breath Res. 2013, 7, 037106. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Instrumentation | Biological Sample | Method Validation | Metabolites Coverage and RT (min) | Data Processing Strategies | Ref |
|---|---|---|---|---|---|
| UHPLC-Q-TOF-MS and UHPLC-QTRAP-MS | Human urine | 69% and 94% of metabolites displayed a relative standard deviation (RSD) of <10% and <20%, respectively. After normalization and internal standard calibration, 94% and 97% of metabolites had an RSD of < 20%, respectively. | 419 compounds in electrospray ionization quadrupole (ESI) positive mode and 449 compounds in ESI negative mode were detected. RT = 26 min. | Analyst 1.6 software (AB SCIEX, USA) was used to conduct peak integration. The non-targeted analysis data were imported to the SIEVE software (ThermoFisher, USA) package to extract the metabolite features. | [40] |
| LC-QTRAP-MS | Human urine | The slopes of linear regressions were approximately 1.00 (1.0291). RSDs were less than 9.0% with a correlation coefficient (R2) of 0.9998. | The number of accurately quantified metabolites from 103 thiols, increased from 64 (62%) to 99 (96%). RT = 55 min. | Manual data acquisition and processing through Bruker Daltonics Control 3.4 and Bruker Daltonics Data analysis 4.0 software (Bruker Daltonics, Bremen, Germany). | [41] |
| GC-MS | Human urine | 75.7%, 89.5%, and 95.9% of the peaks had RSDs of <10%, 20%, and 30%, respectively. The intraday RSDs for 80.2%, 91.6%, and 95.1% of the peaks were <10%, 20%, and 30%, respectively. The interday RSD for 64.3%, 86.9%, and 91.6% of the peaks were <10%, 20%, and 30%, respectively. | A total of 76 differential metabolites were defined, 58 of which were verified. RT = 30 min. | Changes in the levels of the differential metabolites were visualized with MultiExperiment Viewer (http://www.tm4.org). The altered pathways were determined with MetaboAnalyst 2.0 (http://www.metaboanalyst.ca). | [52] |
| UHPLC-Q-Orbitrap-MS and UHPLC-QTRAP-MS | Human urine | The RSDs of all the standards were below 10%. 94% and 80% of the peaks had RSDs of <30% and <20% in the QC samples, respectively. The 95.5% and 90% peaks had RSDs of <30% and <20% in the urine samples, respectively. | 780 metabolites were defined. A total of 48 metabolites were chosen, 26 of which were identified. RT = 28 min. | Ion-pairs selection was from two sources: (1) untargeted metabolic analysis; (2) key metabolites in the metabolic pathways chosen from free databases. MultiQuant software (Applied Biosystem/MDS Sciex, Carlsbad, CA) was used to extract the peaks. | [53] |
| UHPLC-Q-TOF-MS and UHPLC-QQQ-MS | Human serum | 34% of the detected metabolites had an RSD < 5% and 76% had an RSD < 10%. | 518 ion-pairs were defined for subsequent MRM detection. RT = 29.9 min. | Manual peak detection and alignment through XCMS software. | [37] |
| LC-QTOF-MS | Human serum | The minimum absolute height required was set at 3000 counts, which was also used for 100% of analyses in the recursive step to minimize the experimental variability. | 139 and 158 molecular entities in the negative and positive ionization mode, respectively, were obtained. RT = 20 min. | The MassHunter Workstation software package was used to process all the data obtained by LC/Q-TOF in the MS/MS mode. | [38] |
| UHPLC-Q-TOF-MS and UHPLC-QQQ-MS | Human serum | 91% of contents had an RSD of <30% from the QC samples. | 1446 metabolite MRM transitions in the MRM-Ion Pair Finder. RT = 23 min. | Efficient data processing strategy, with the processing time markedly shortened by the homemade MRM-Ion Pair Finder software. | [31] |
| LC−QQQ-MS | Human serum | The linearity r at 0.82 ± 0.26 of amino acids. >40% of the GOT-MS MRMs had CVs < 5%; the average CV of the detected metabolites was at 7.8 ± 7.0%. | 595 precursor ions and 1890 multiple reaction monitoring transitions (MRM). RT = 9 min. | Manual inspection based on symmetry, peak width, and MS peak area extraction by Agilent MassHunter Qualitative Analysis and Quantitative Analysis software (Agilent Technologies, Inc., Santa Clara, CA). | [24] |
| UHPLC-Q-TOF-MS and UHPLC/QTRAP-MS | Human serum | The change folds of a peak area between these two QC samples ranged from 0.002 to 14. 767 (94.3%) and 759 (93.3%) of the peaks had a CV of <30% in the b-QC and p-QC replicates, respectively. | 813 ions were steadily detected in the QC samples. RT = 30 min. | The integrations of the peak areas were processed by the software provided by the instrument vendor. The efficient data processing strategy used the MRM-Ion Pair Finder software. | [28] |
| UHPLC-QTRAP-MS and LC-MS | Human serum | For the validation set, the AUC was from 0.676 to 0.875. Sensitivity was from 0.504 to 0.921. Specificity was from 0.528 to 0.784. | 239 metabolites were identified. RT = 20 min. | Manual peak detection and alignment through the XCMS software. Home-developed database and online databases (HMDB and Metlin) or confirming with authentic standards were used. | [54] |
| 2 D-UHPLC-QTRAP-MS | Human plasma | The linear regression r varied between 0.9902 and 0.9993. The average accuracies for the standard samples were between 0.11% and 13.90%, and the average intra-/inter-day precisions of the standard samples were between 0.66% and 16.46%. The intra-/inter-day precisions in the complex plasma matrix were between 2.05% and 19.49%. | 78 metabolites were confidently confirmed, from which 73 metabolites can be accurately quantified. Untargeted profiling of 4651 features of high reliability and validity was achieved. RT = 27 min. | Targeted qualitative and quantitative was performed using the TraceFinder 3.3 software (Thermo Fisher Scientific, Waltham, MA). All the calibration curves were linear and weighted 1/x. The untargeted analysis was performed by the Progenesis QI 2.0 software (Waters, Milford, MA, USA). | [26] |
| UHPLC-Q-Orbitrap-MS and UHPLC-Qtrap-MS | Human plasma | 43 yielded good linear functions (R2 > 0.99) had RSDs lower than 20% and an accuracy between 80% and 120%. | 1658 characteristic ion-pairs from 1324 metabolites. RT = 35 min. | 48 metabolites established DITQM; manually generate “one feature for one peak” metabolomics data, further confirmed through the extracted ion chromatograms (XICs). | [39] |
| UHPLC-Q-TOF-MS and UHPLC-QTRAP-MS | Human plasma | The intraday RSDs of 82.4% of the metabolites were <15%. 25.1% of the metabolites had RSD values of >15%. | A total of 1373 unique metabolite ion-pairs were obtained in the positive ion mode. 162 significantly changed metabolites were defined. RT = 30 min. | Peak detection and alignment were performed by the MarkerView software (AB SCIEX, Framingham, USA) MS-DIAL software and homemade C-package were used to handle MS2. UHPLC-MRM MS data were disposed of in Analyst 1.6 software (AB SCIEX, Framingham, USA). | [55] |
| UHPLC-QQQ-MS | Human plasma | The optimized MRMs had intensities of >1000 and signal-to-noise-ratios (S/Ns) of >3. The intraday and interday median CVs were 4.86% and 8.79%, respectively. The median r was 0.96. | 927 metabolites were measured. 310 were confirmed using pure chemical standards, while the rest were annotated by identification level using database entries. RT = 15 min. | The entire LC-MS system was controlled by the Mass Hunter Workstation software (Agilent, Santa Clara, CA). The extracted MRM peaks were integrated using the Mass Hunter Quantitative Data Analysis software. | [51] |
| UHPLC-Q-TOF-MS | Mouse serum | 66.7% of metabolites displayed an RSD of <10%. 99.8% of metabolites had an RSD of <20%. After normalization by the total intensity and sample median, 99.8% and 99.4% of metabolites exhibited an RSD of <20%, respectively | 2081 ions were obtained after data filtering. 569 peaks were selected to perform tsMIM-based pseudotargeted analysis. RT = 26 min. | The raw data were extracted and aligned by the Progenesis QI software. The peak areas were collected by the TargetLynx software. | [43] |
| UHPLC-Q-TOF-MS | Mouse plasma | 97.4% and 95.4% of the selected ions in the positive and negative ion modes have RSD values of less than 20%, respectively. | 1423 and 1141 ions were generated in the positive and negative ion modes, respectively. RT = 27 min. | The raw data files were uploaded into the Progenesis QI software to perform chromatographic peak alignment, data normalization, and peak picking using the default settings. MassLynx XS (Waters Corp., Manchester, UK) was used for the targeted extraction. The selected features were tentatively annotated. | [42] |
| UHPLCQ-TOF-MS | Mouse plasma | Almost 90% of the tsMIM- detected metabolites had an RSD of <20%. 42% of metabolites had RDSs of <5%. | 387 ions were detected. 17 metabolites were selected as biomarkers. RT = 26 min. | The peak areas of the metabolites were processed using the TargetLynx software. | [27] |
| UHPLC-Q-Orbitrap-MS and UHPLC-QQQ MS | Mouse brain and liver, cancer cells, and human plasma | The linear of IS r > 0.99. The intraday RSDs were from 0.8% to 4.3%; the inter-day RSDs were from 2.4% to 16.8%. 51% and 94% of the detected lipids had RSDs of <5% and 20% in positive mode and 94% had RSDs of <10% in negative mode. | A total of 3377 targeted lipid ion pairs with over 7000 lipid molecular structures were defined. RT = 20 min. | The raw data were processed with Analyst software. Normalization of the lipids by the appropriate lipid ISs. | [44] |
| UHPLC-Q-TOF-MS and UHPLC-QTRAP-MS | Rice seed | 89.5% of peaks had RSDs < 20%. The linear range was 2.5–4 orders of magnitude, and the r was in the range 0.996–0.999. The recoveries were 85.9–106.3% for positive mode and 73.3–98.2% for negative mode, respectively. | A total of 749 and 617 ion pairs in the positive and negative modes were achieved, respectively. Among them, about 200 metabolites were identified or tentatively identified. RT = 30 min. | All the ions were extracted by the Analyst software. Zero values were removed by the 80% rule. | [28] |
| GC-MS | Commercial cigarettes | >81.2% of peaks had RSDs of <20%. | 312 components and their related quantitation ions. A total of 90 compounds were elucidated. RT = 72.5 min. | The integration of the chromatography peaks was performed using the Agilent MSD ChemStation (Agilent Technologies). Peak areas of all the components were divided by those of the internal standard and then scaled to zero mean and unit variance. | [36] |
| GC-MS | Tobacco leaf | 47.3% of components had an r of >0.99; 36% of components had an RSD of <5%; 93% of components had an RSD of < 20%. | 167 differential components (p < 0.05) were screened out. RT = 72.5 min. | The acquired GC/MS raw data of the QC sample were imported into the AMDIS software (version 2.62, NIST, USA) for peak deconvolution and detection. The quantitative ion selection algorithm was written in Visual C++ (version 6.0, Microsoft). The component peak area was divided by that of the internal standard. | [25] |
| SESI-QQQ-MS | Bacterial culture | 47% of features showed an r of >0.90. 65.9% of features had a CV of <20% | 75 features in the SESI-GOT-MS/MS panel were established. RT = 1 min. | Manual inspection based on symmetry, peak width, and MS peak area extraction by Agilent MassHunter Qualitative Analysis and Quantitative Analysis software. | [29] |
| UHPLC-QQQ-MS | Bacterial culture | 54.9% of peaks had measurement CVs of <10%; 0.84% had CVs of >40%. 51.97% of peaks had CVs of <10%; 0% had CVs of >40%. | A total of 464 metabolite peaks were detected. RT = 20 min. | All the raw metabolomics data were inspected by the Quanbrowser module of Xcalibur version 4.0 (Thermo Fisher Scientific) and the Thermo TSQ LC-SIM Data Processor (V0.1.35 Engineering Sample). | [45] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Zhong, F.; Zhu, J. Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches. Metabolites 2020, 10, 348. https://doi.org/10.3390/metabo10090348
Chen L, Zhong F, Zhu J. Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches. Metabolites. 2020; 10(9):348. https://doi.org/10.3390/metabo10090348
Chicago/Turabian StyleChen, Li, Fanyi Zhong, and Jiangjiang Zhu. 2020. "Bridging Targeted and Untargeted Mass Spectrometry-Based Metabolomics via Hybrid Approaches" Metabolites 10, no. 9: 348. https://doi.org/10.3390/metabo10090348