Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
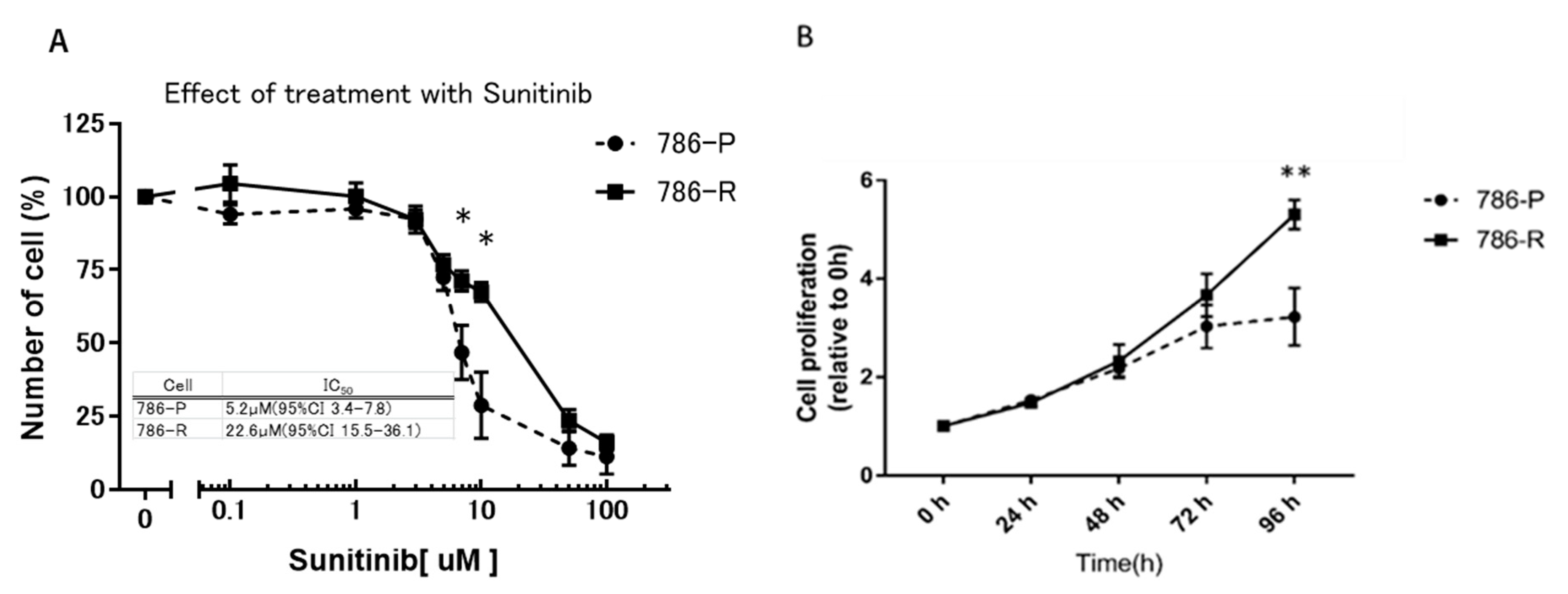
2.1. Comparison of Growth Inhibitory Effect of Sunitinib in Cell Lines
2.2. Establishment of a Sunitinib-Resistant 786-O Cell Line (786-R) for In Vitro and In Vivo Examination of Sunitinib Resistance
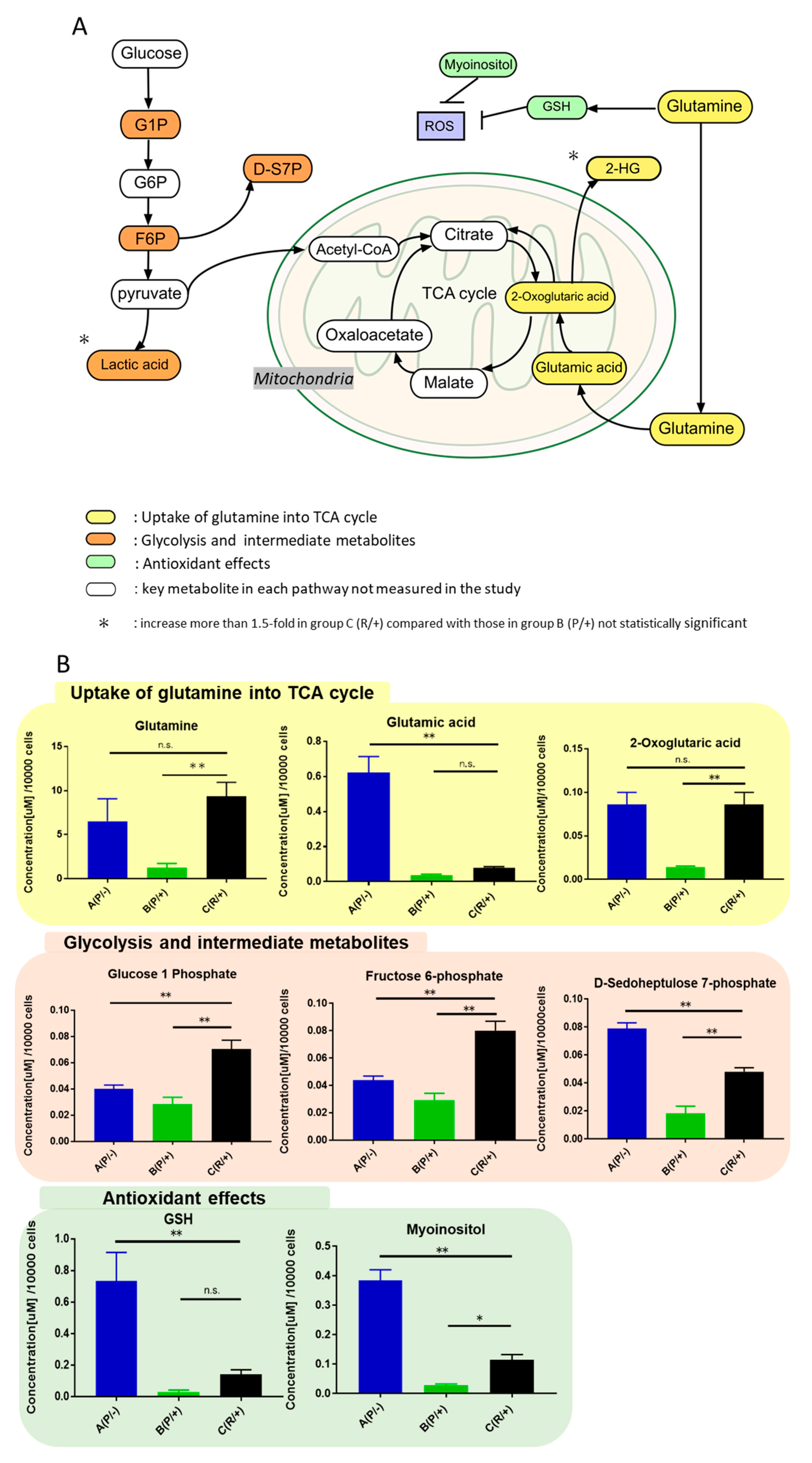
2.3. Identification of Upregulated Metabolites in Sunitinib-Resistant Cells
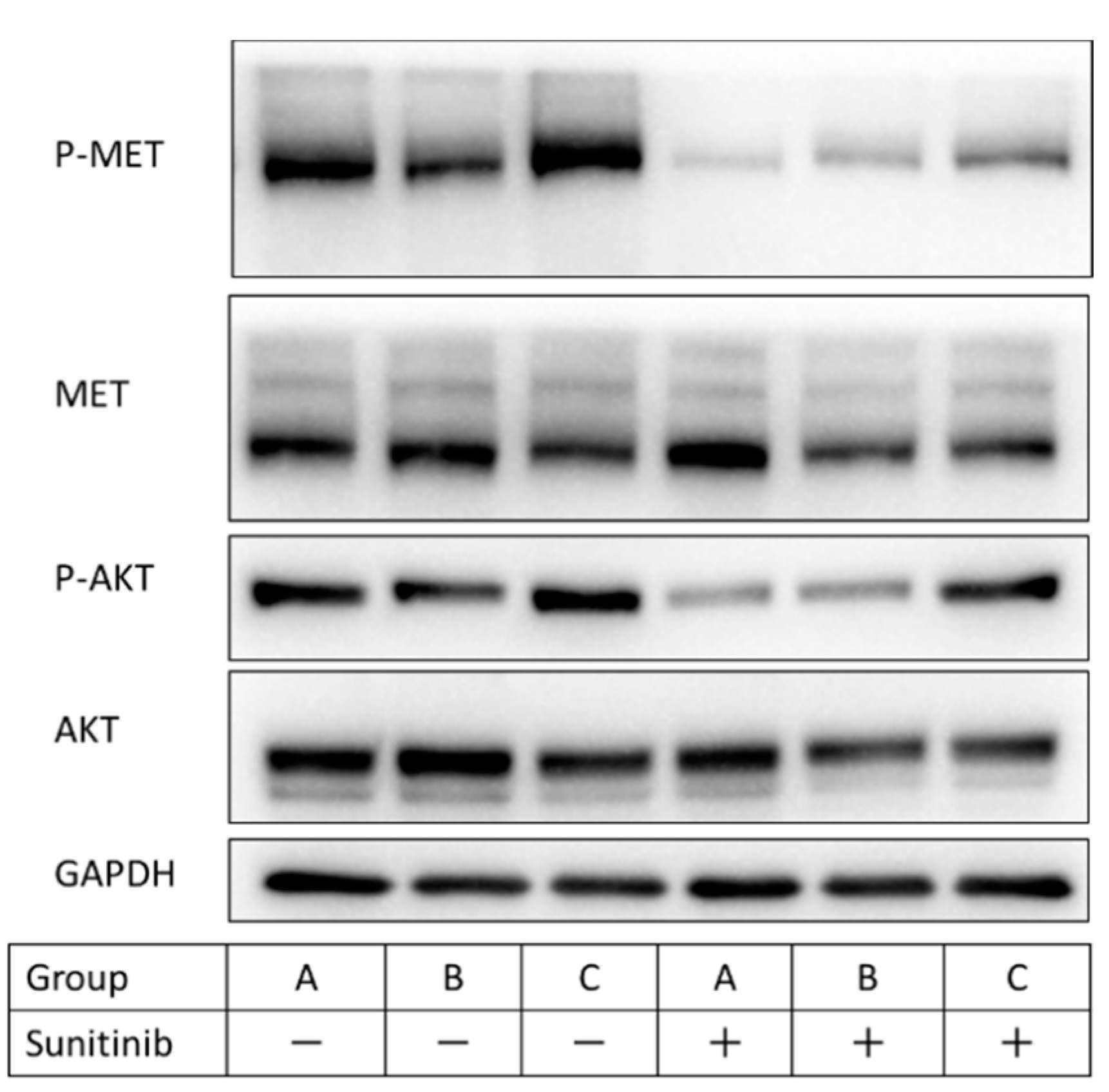
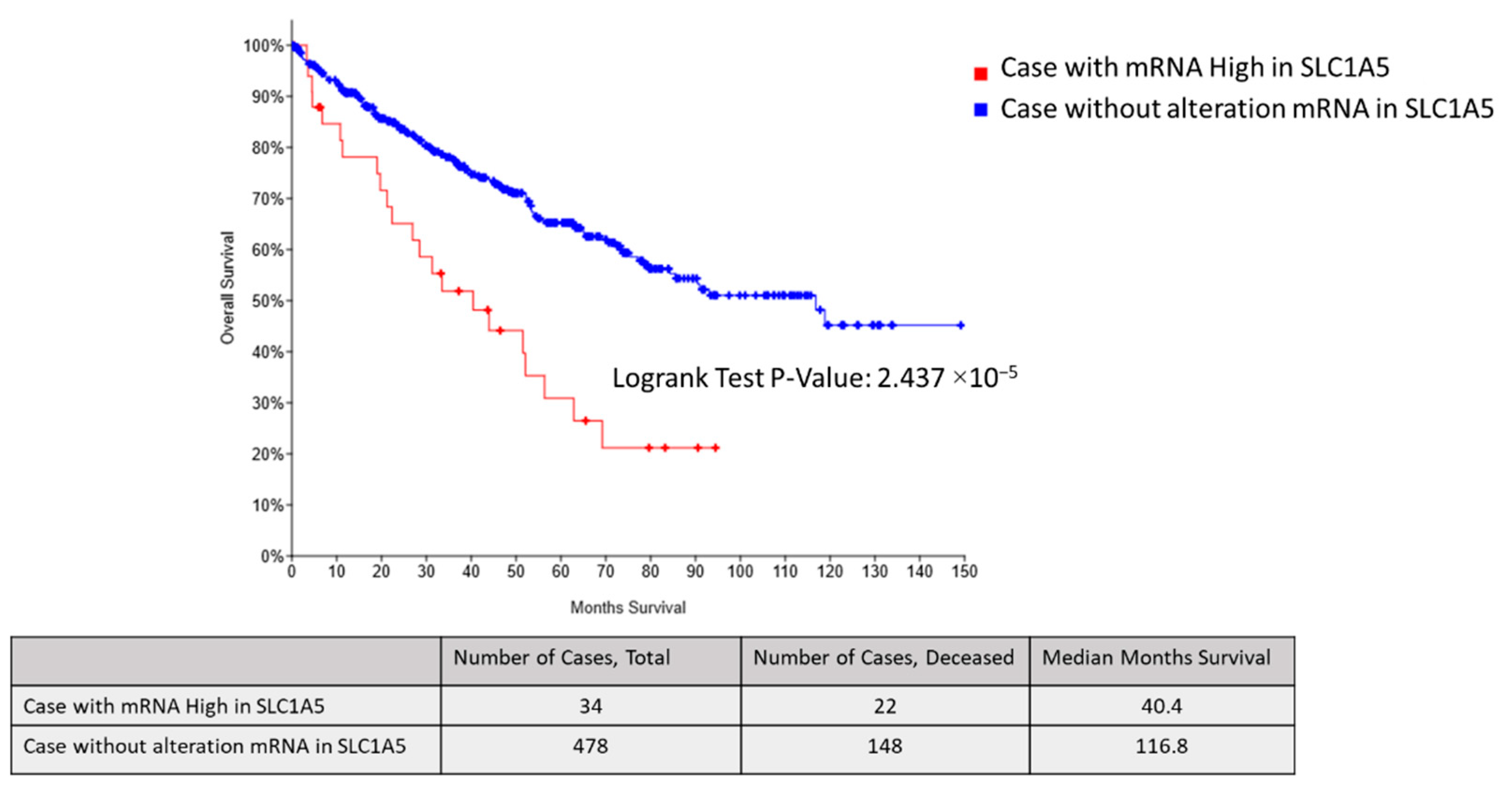
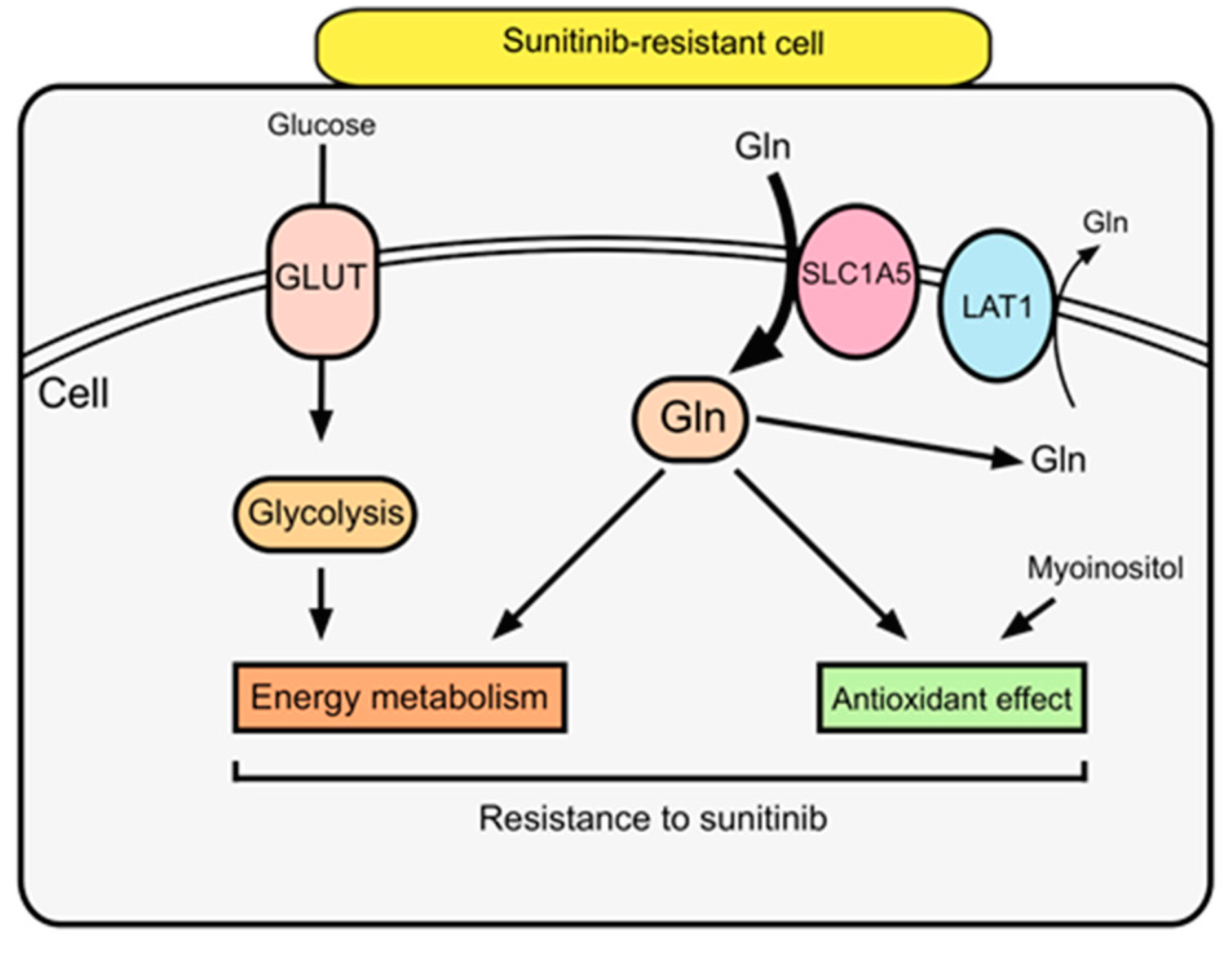
2.4. SLC1A5 and LAT1 Expression Related to Glutamine Uptake in Sunitinib-Resistant Cells
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Lines and Culture
4.3. WST Assay
4.4. Cell Proliferation Assay
4.5. Establishment of Sunitinib-Resistant Mouse Model
4.6. Wound-Healing Assay
4.7. Two-Chamber Assay
4.8. Phosphorylation Stimulation and Antibodies
4.9. Western Blot Analysis
4.10. Chemicals and Reagents for LC-MS/MS Analysis
4.11. LC-MS/MS Conditions
4.12. Sample Preparation for LC-MS/MS
4.13. RNA Extraction and Quantitative RT-PCR
4.14. Bioinformatics Analysis
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cairns, P. Renal cell carcinoma. Cancer Biomark. Sect. A Dis. Markers 2010, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Matsuda, T.; Shibata, A.; Katanoda, K.; Sobue, T.; Nishimoto, H. Cancer incidence and incidence rates in Japan in 2009: A study of 32 population-based cancer registries for the Monitoring of Cancer Incidence in Japan (MCIJ) project. Jpn. J. Clin. Oncol. 2015, 45, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, M.; Larkin, J.; Ravaud, A. Experience with sunitinib in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2012, 4, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Abrams, T.J.; Lee, L.B.; Murray, L.J.; Pryer, N.K.; Cherrington, J.M. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol. Cancer Ther. 2003, 2, 471–478. [Google Scholar]
- Goto, Y.; Kurozumi, A.; Nohata, N.; Kojima, S.; Matsushita, R.; Yoshino, H.; Yamazaki, K.; Ishida, Y.; Ichikawa, T.; Naya, Y.; et al. The microRNA signature of patients with sunitinib failure: Regulation of UHRF1 pathways by microRNA-101 in renal cell carcinoma. Oncotarget 2016, 7, 59070–59086. [Google Scholar] [CrossRef] [Green Version]
- Haouala, A.; Rumpold, H.; Untergasser, G.; Buclin, T.; Ris, H.B.; Widmer, N.; Decosterd, L.A. siRNA-mediated knock-down of P-glycoprotein expression reveals distinct cellular disposition of anticancer tyrosine kinases inhibitors. Drug Metab. Lett. 2010, 4, 114–119. [Google Scholar] [CrossRef]
- Hu, S.; Chen, Z.; Franke, R.; Orwick, S.; Zhao, M.; Rudek, M.A.; Sparreboom, A.; Baker, S.D. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6062–6069. [Google Scholar] [CrossRef] [Green Version]
- Vermeersch, K.A.; Styczynski, M.P. Applications of metabolomics in cancer research. J. Carcinog. 2013, 12, 9. [Google Scholar] [CrossRef]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Palmnas, M.S.; Vogel, H.J. The future of NMR metabolomics in cancer therapy: Towards personalizing treatment and developing targeted drugs? Metabolites 2013, 3, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, A.M.; de Pinho, P.G.; Monteiro, M.S.; Duarte, I.F. NMR metabolomics of renal cancer: An overview. Bioanalysis 2015, 7, 2361–2374. [Google Scholar] [CrossRef] [PubMed]
- Zira, A.N.; Theocharis, S.E.; Mitropoulos, D.; Migdalis, V.; Mikros, E. (1)H NMR metabonomic analysis in renal cell carcinoma: A possible diagnostic tool. J. Proteome Res. 2010, 9, 4038–4044. [Google Scholar] [CrossRef]
- Di Napoli, A.; Signoretti, S. Tissue biomarkers in renal cell carcinoma: Issues and solutions. Cancer 2009, 115, 2290–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maria, R.M.; Altei, W.F.; Selistre-de-Araujo, H.S.; Colnago, L.A. Impact of chemotherapy on metabolic reprogramming: Characterization of the metabolic profile of breast cancer MDA-MB-231 cells using (1)H HR-MAS NMR spectroscopy. J. Pharm. Biomed. Anal. 2017, 146, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Kawasaki, Y.; Maekawa, M.; Takasaki, S.; Saigusa, D.; Ota, H.; Shimada, S.; Yamashita, S.; Mitsuzuka, K.; Yamaguchi, H.; et al. Value of global metabolomics in association with diagnosis and clinicopathological factors of renal cell carcinoma. Int. J. Cancer 2019, 145, 484–493. [Google Scholar] [CrossRef]
- Saigusa, D.; Okamura, Y.; Motoike, I.N.; Katoh, Y.; Kurosawa, Y.; Saijyo, R.; Koshiba, S.; Yasuda, J.; Motohashi, H.; Sugawara, J.; et al. Establishment of Protocols for Global Metabolomics by LC-MS for Biomarker Discovery. PLoS ONE 2016, 11, e0160555. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [Green Version]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic Routes in Inflammation: The Citrate Pathway and its Potential as Therapeutic Target. Curr. Med. Chem. 2019, 26, 7104–7116. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 2016, 1863, 2394–2412. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Deng, L. Sphingosine kinase-1 activation causes acquired resistance against Sunitinib in renal cell carcinoma cells. Cell Biochem. Biophys. 2014, 68, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Sakai, I.; Miyake, H.; Fujisawa, M. Acquired resistance to sunitinib in human renal cell carcinoma cells is mediated by constitutive activation of signal transduction pathways associated with tumour cell proliferation. BJU Int. 2013, 112, E211–E220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, H.; Fujiwara, T.; Sato, H.; Terui, A.; Hisaka, A. Investigation of Metabolomic Changes in Sunitinib-Resistant Human Renal Carcinoma 786-O Cells by Capillary Electrophoresis-Time of Flight Mass Spectrometry. Biol. Pharm. Bull. 2018, 41, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.M.; Darzi, A.W.; Takats, Z.; Lindon, J.C. Metabolic phenotyping in clinical and surgical environments. Nature 2012, 491, 384–392. [Google Scholar] [CrossRef]
- Busch, J.; Seidel, C.; Weikert, S.; Wolff, I.; Kempkensteffen, C.; Weinkauf, L.; Hinz, S.; Magheli, A.; Miller, K.; Grunwald, V. Intrinsic resistance to tyrosine kinase inhibitors is associated with poor clinical outcome in metastatic renal cell carcinoma. BMC Cancer 2011, 11, 295. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.A.; Hutson, T.E.; Elson, P.; Cowey, C.L.; Gilligan, T.; Nemec, C.; Dreicer, R.; Bukowski, R.M.; Rini, B.I. Sorafenib in patients with metastatic renal cell carcinoma refractory to either sunitinib or bevacizumab. Cancer 2010, 116, 5383–5390. [Google Scholar] [CrossRef]
- Bost, F.; Decoux-Poullot, A.G.; Tanti, J.F.; Clavel, S. Energy disruptors: Rising stars in anticancer therapy? Oncogenesis 2016, 5, e188. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, H.; Enokida, H.; Itesako, T.; Kojima, S.; Kinoshita, T.; Tatarano, S.; Chiyomaru, T.; Nakagawa, M.; Seki, N. Tumor-suppressive microRNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer Sci. 2013, 104, 1567–1574. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Gibney, G.T.; Aziz, S.A.; Camp, R.L.; Conrad, P.; Schwartz, B.E.; Chen, C.R.; Kelly, W.K.; Kluger, H.M. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 343–349. [Google Scholar] [CrossRef]
- Miyata, Y.; Kanetake, H.; Kanda, S. Presence of phosphorylated hepatocyte growth factor receptor/c-Met is associated with tumor progression and survival in patients with conventional renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 4876–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Hoerner, C.R.; Chen, V.J.; Fan, A.C. The ‘Achilles Heel’ of Metabolism in Renal Cell Carcinoma: Glutaminase Inhibition as a Rational Treatment Strategy. Kidney Cancer 2019, 3, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rives, M.L.; Javitch, J.A.; Wickenden, A.D. Potentiating SLC transporter activity: Emerging drug discovery opportunities. Biochem. Pharmacol. 2017, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Petrashen, A.P.; Sanders, J.A.; Peterson, A.L.; Sedivy, J.M. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell 2019, 18, e12947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H.; Nohata, N.; Miyamoto, K.; Yonemori, M.; Sakaguchi, T.; Sugita, S.; Itesako, T.; Kofuji, S.; Nakagawa, M.; Dahiya, R.; et al. PHGDH as a Key Enzyme for Serine Biosynthesis in HIF2α-Targeting Therapy for Renal Cell Carcinoma. Cancer Res. 2017, 77, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; An, H.; Chang, Y.; Zhang, W.; Zhu, Y.; Xu, L.; Xu, J. High expression of Solute Carrier Family 1, member 5 (SLC1A5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci. Rep. 2015, 5, 16954. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Ren, M.; Zeng, X.; He, P.; Ma, X.; Qiao, S. Leucine stimulates ASCT2 amino acid transporter expression in porcine jejunal epithelial cell line (IPEC-J2) through PI3K/Akt/mTOR and ERK signaling pathways. Amino Acids 2014, 46, 2633–2642. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Y.; Zhao, L.; Li, Y.; Zheng, J. Glutaminase 1 is essential for the differentiation, proliferation, and survival of human neural progenitor cells. Stem Cells Dev. 2014, 23, 2782–2790. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, T.; Kawasaki, Y.; Maekawa, M.; Takasaki, S.; Morozumi, K.; Sato, M.; Shimada, S.; Kawamorita, N.; Yamashita, S.; Mitsuzuka, K.; et al. Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma. Metabolites 2021, 11, 1. https://doi.org/10.3390/metabo11010001
Sato T, Kawasaki Y, Maekawa M, Takasaki S, Morozumi K, Sato M, Shimada S, Kawamorita N, Yamashita S, Mitsuzuka K, et al. Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma. Metabolites. 2021; 11(1):1. https://doi.org/10.3390/metabo11010001
Chicago/Turabian StyleSato, Tomonori, Yoshihide Kawasaki, Masamitsu Maekawa, Shinya Takasaki, Kento Morozumi, Masahiko Sato, Shuichi Shimada, Naoki Kawamorita, Shinichi Yamashita, Koji Mitsuzuka, and et al. 2021. "Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma" Metabolites 11, no. 1: 1. https://doi.org/10.3390/metabo11010001
APA StyleSato, T., Kawasaki, Y., Maekawa, M., Takasaki, S., Morozumi, K., Sato, M., Shimada, S., Kawamorita, N., Yamashita, S., Mitsuzuka, K., Mano, N., & Ito, A. (2021). Metabolomic Analysis to Elucidate Mechanisms of Sunitinib Resistance in Renal Cell Carcinoma. Metabolites, 11(1), 1. https://doi.org/10.3390/metabo11010001