
Physiological and Pathological Roles of Aldose Reductase



Abstract
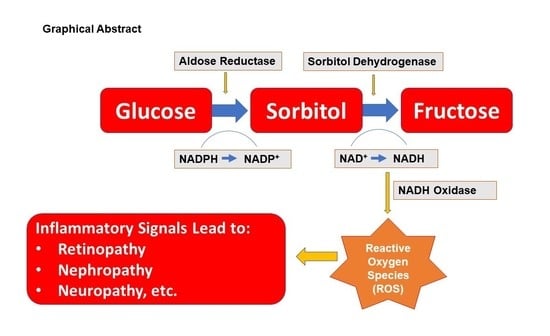
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytotoxic Compound | Km (mM) | kcat (min−1) | kcat/Km (min−1·µM−1) | Reference(s) |
|---|---|---|---|---|
| Acrolein (Figure 4a) | 0.80 ± 0.21 | 37.6 | 47 M−1 min−1 | [14] |
| 4HNE (Figure 4b) | 22 | 102 | 4.6 × 106 M−1 min−1 | [15] |
| Glyoxal (Figure 4c) | 514 | 154 | 3.0 × 105 M−1 min−1 | [15] |
| Methylglyoxal (Figure 4d) | 0.008 | 142 | 1.8 × 107 M−1 min−1 | [15,16] |
| ONE (Figure 4e) | 0.0042 ± 0.0046 | 92.2 ± 3.55 | 2190 ± 120 M−1 min−1 | [17] |
| Glucose (Figure 4f) | 0.68 | 0.15 | 9.1 × 102 M−1 min−1 | [16,18,19] |
| Galactose (Figure 4g) | 21 | 222 | 10.57 M−1 min−1 | [20] |
1.1. Structure of Aldose Reductase
1.2. Functional Aspects of Aldose Reductase
1.3. Role of Aldose Reductase in Human Diseases
1.3.1. Diabetes
1.3.2. Cardiovascular Diseases
1.4. Asthma
1.5. Oculopathy
1.6. Nephropathy
1.7. Neuropathy
1.8. Sepsis
1.9. Cancer
2. Concluding Remarks, and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blakeley, M.P.; Ruiz, F.; Cachau, R.; Hazemann, I.; Meilleur, F.; Mitschler, A.; Ginell, S.; Afonine, P.; Ventura, O.N.; Cousido-Siah, A.; et al. Quantum Model of Catalysis Based on a Mobile Proton Revealed by Subatomic X-ray and Neutron Diffraction Studies of H-Aldose Reductase. Proc. Natl. Acad. Sci. USA 2008, 105, 1844–1848. [Google Scholar] [CrossRef]
- Wilson, D.; Bohren, K.; Gabbay, K.; Quiocho, F. An Unlikely Sugar Substrate Site in the 1.65 a Structure of the Human Aldose Reductase Holoenzyme Implicated in Diabetic Complications. Science 1992, 257, 81–84. [Google Scholar] [CrossRef]
- Umegaki, H.; Hayashi, T.; Nomura, H.; Yanagawa, M.; Nonogaki, Z.; Nakshima, H.; Kuzuya, M. Cognitive Dysfunction: An Emerging Concept of a New Diabetic Complication in the Elderly. Geriatr. Gerontol. Int. 2013, 13, 28–34. [Google Scholar] [CrossRef]
- Leslie, R. United Kingdom Prospective Diabetes Study (UKPDS): What Now or So What? Diabetes Metab. Res. Rev. 1999, 15, 65–71. [Google Scholar] [CrossRef]
- Ferreira, F.N.; Crispim, D.; Canani, L.H.; Gross, J.L.; dos Santos, K.G. Association Study of Sorbitol Dehydrogenase-888G> C Polymorphism with Type 2 Diabetic Retinopathy in Caucasian-Brazilians. Exp. Eye Res. 2013, 115, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Akileshwari, C.; Muthenna, P.; Nastasijević, B.; Joksić, G.; Petrash, J.M.; Reddy, G.B. Inhibition of Aldose Reductase by Gentiana lutea Extracts. Exp. Diabetes Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. The Aldose Reductase Controversy. Diabetes 1994, 43, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.A.; Lattimer, S.A.; Sima, A.A. Sorbitol, Phosphoinositides, and Sodium-Potassium-ATPase in the Pathogenesis of Diabetic Complications. N. Engl. J. Med. 1987, 316, 599–606. [Google Scholar] [CrossRef]
- Hotta, N. New Concepts and Insights on Pathogenesis and Treatment of Diabetic Complications: Polyol Pathway and Its Inhibition. Nagoya J. Med. Sci. 1997, 60, 89–100. [Google Scholar] [PubMed]
- Cowell, R.M.; Russell, J.W. Nitrosative Injury and Antioxidant Therapy in the Management of Diabetic Neuropathy. J. Investig. Med. 2004, 52, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension: Clinical Implications and Therapeutic Possibilities. Diabetes Care 2008, 31, 170–180. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Oxidative and Reductive Metabolism of Lipid-Peroxidation Derived Carbonyls. Chem. Biol. Interact. 2015, 234. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Yadav, U.C.; Reddy, A.B.; Saxena, A.; Tammali, R.; Shoeb, M.; Ansari, N.H.; Bhatnagar, A.; Petrash, M.J.; Srivastava, S.; et al. Aldose Reductase Inhibition Suppresses Oxidative Stress-Induced Inflammatory Disorders. Chem. Biol. Interact. 2011, 191, 330–338. [Google Scholar] [CrossRef]
- Srivastava, S.; Watowich, S.J.; Petrash, J.M.; Srivastava, S.K.; Bhatnagar, A. Structural and Kinetic Determinants of Aldehyde Reduction by Aldose Reductase. Biochemistry 1999, 38, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L.; Kolb, N.S.; Vander Jagt, T.J.; Chino, J.; Martinez, F.J.; Hunsaker, L.A.; Royer, R.E. Substrate Specificity of Human Aldose Reductase: Identification of 4-Hydroxynonenal as an Endogenous Substrate. Biochim. Biophys. Acta 1995, 1249, 117–126. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Robinson, B.; Taylor, K.K.; Hunsaker, L.A. Reduction of Trioses by NADPH-Dependent Aldo-Keto Reductases. Aldose Reductase, Methylglyoxal, and Diabetic Complica-Tions. J. Biol. Chem. 1992, 267, 4364–4369. [Google Scholar] [CrossRef]
- Doorn, J.A.; Srivastava, S.K.; Petersen, D.R. Aldose Reductase Catalyzes Reduction of the Lipid Peroxidation Product 4-Oxonon-2-Enal. Chem. Res. Toxicol. 2003, 16, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Hair, G.A.; Das, B. Activated and Unactivated Forms of Human Erythrocyte Aldose Reductase. Proc. Natl. Acad. Sci. USA 1985, 82, 7222–7226. [Google Scholar] [CrossRef]
- Bohren, K.M.; Grimshaw, C.E.; Gabbay, K.H. Catalytic Effectiveness of Human Aldose Reductase. Critical Role of C-Terminal Domain. J. Biol. Chem. 1992, 267, 20965–20970. [Google Scholar] [CrossRef]
- Cao, D.; Fan, S.T.; Chung, S.S. Identification and Characterization of a Novel Human Aldose Reductase-Like Gene. J. Biol. Chem. 1998, 273, 11429–11435. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Manna, P.; Gachhui, R.; Sil, P.C. D-Saccharic Acid 1,4-Lactone Protects Diabetic Rat Kidney by Ameliorating Hyperglycemia-Mediated Oxidative Stress and Renal Inflammatory Cytokines via NF-κB and PKC Signaling. Toxicol. Appl. Pharmacol. 2013, 267, 16–29. [Google Scholar] [CrossRef]
- Ciddi, V.; Dodda, D. Therapeutic Potential of Resveratrol in Diabetic Complications: In Vitro and In Vivo Studies. Pharmacol. Rep. PR 2014, 66, 799–803. [Google Scholar] [CrossRef]
- Thiagarajan, R.; Varsha, M.; Srinivasan, V.; Ravichandran, R.; Saraboji, K. Vitamin K1 Prevents Diabetic Cataract by Inhibiting Lens Aldose Reductase 2 (ALR2) Activity. Sci. Rep. 2019, 9, 14684. [Google Scholar] [CrossRef]
- Caglayan, C.; Demir, Y.; Kucukler, S.; Taslimi, P.; Kandemir, F.M.; Gulçin, İ. The Effects of Hesperidin on Sodium Arsenite-Induced Different Organ Toxicity in Rats on Metabolic Enzymes as Antidiabetic and Anticholinergics Potentials: A Biochemical Approach. J. Food Biochem. 2019, 43, e12720. [Google Scholar] [CrossRef]
- Patil, K.K.; Gacche, R.N. Inhibition of Glycation and Aldose Reductase Activity Using Dietary Flavonoids: A Lens Organ Culture Studies. Int. J. Biol. Macromol. 2017, 98, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Liao, S.; Mi, H.; Guo, C.; Qi, D.; Li, F.; Zhang, C.; Yang, Z. Hesperidin Prevents Retinal and Plasma Abnormalities in Streptozotocin-Induced Diabetic Rats. Molecules 2012, 17, 12868–12881. [Google Scholar] [CrossRef] [PubMed]
- Puppala, M.; Ponder, J.; Suryanarayana, P.; Reddy, G.B.; Petrash, J.M.; LaBarbera, D.V. The Isolation and Characterization of β-Glucogallin as a Novel Aldose Reductase Inhibitor from Emblica officinalis. PLoS ONE 2012, 7, e31399. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Laffin, B.; Ponder, J.; Enzsöly, A.; Németh, J.; LaBarbera, D.V.; Petrash, J.M. Beta-Glucogallin Reduces the Expression of Lipopolysaccharide-Induced Inflammatory Markers by Inhibition of Aldose Reductase in Murine Macrophages and Ocular Tissues. Chem. Biol. Interact. 2013, 202, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.G.; Zhang, L.X.; Liu, X. Progress in Research of Aldose Reductase Inhibitors in Traditional Medicinal Herbs. China J. Chin. Mater. Med. 2005, 30, 1496–1500. [Google Scholar]
- Gupta, S.; Singh, N.; Jaggi, A.S. Alkaloids as Aldose Reductase Inhibitors, with Special Reference to Berberine. J. Altern. Complement. Med. 2014, 20, 195–205. [Google Scholar] [CrossRef]
- Lee, H.S. Rat Lens Aldose Reductase Inhibitory Activities of Coptis japonica Root-Derived Isoquinoline Alkaloids. J. Agric. Food Chem. 2002, 50, 7013–7016. [Google Scholar] [CrossRef]
- Liu, W.; Liu, P.; Tao, S.; Deng, Y.; Li, X.; Lan, T.; Zhang, X.; Guo, F.; Huang, W.; Chen, F.; et al. Berberine Inhibits Aldose Reductase and Oxidative Stress in Rat Mesangial Cells Cultured under High Glucose. Arch. Biochem. Biophys. 2008, 475, 128–134. [Google Scholar] [CrossRef]
- Liu, W.H.; Hei, Z.Q.; Nie, H.; Tang, F.T.; Huang, H.Q.; Li, X.J.; Deng, Y.H.; Chen, S.R.; Guo, F.F.; Huang, W.G.; et al. Berberine Ameliorates Renal Injury in Streptozotocin-Induced Diabetic Rats by Suppression of Both Oxidative Stress and Aldose Reductase. Chin. Med. J. 2008, 121, 706–712. [Google Scholar] [CrossRef]
- Yang, H.Z.; Zhou, M.M.; Zhao, A.H.; Xing, S.N.; Fan, Z.Q.; Jia, W. Study on Effects of Baicalin, Berberine and Astragalus Polysaccharides and Their Combinative Effects on Aldose Reductase In Vitro. J. Chin. Med. Mater. 2009, 32, 1259–1261. [Google Scholar]
- Qiu, L.; Guo, C. Natural Aldose Reductase Inhibitor: A Potential Therapeutic Agent for Non-alcoholic Fatty Liver Disease. Curr. Drug Targets 2020, 21, 599–609. [Google Scholar] [CrossRef]
- Qiu, L.; Guo, C.; Hua, B. Aldose Reductase Inhibitors of Plant Origin in the Prevention and Treatment of Alcoholic Liver Disease: A Minireview. Biomed. Res. Int. 2019, 2019, 3808594. [Google Scholar] [CrossRef] [PubMed]
- Veeresham, C.; Rama Rao, A.; Asres, K. Aldose Reductase Inhibitors of Plant Origin. Phytother. Res. PTR 2014, 28, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, S.; Cui, Z.; Nie, H.; Han, D.; Yan, H. Screening and Isolating Major Aldose Reductase Inhibitors from the Seeds of Evening Primrose (Oenothera biennis). Molecules 2019, 24, 2709. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Yoon, N.Y.; Bae, H.J.; Min, B.S.; Choi, J.S. Inhibitory Activities of the Alkaloids from Coptidis Rhizoma against Aldose Reductase. Arch. Pharmacal Res. 2008, 31, 1405–1412. [Google Scholar] [CrossRef]
- Ran, Q.; Wang, J.; Wang, L.; Zeng, H.R.; Yang, X.B.; Huang, Q.W. Rhizoma coptidis as a Potential Treatment Agent for Type 2 Diabetes mellitus and the Underlying Mechanisms: A Review. Front. Pharmacol. 2019, 10, 805. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, X.; Zhang, X.; Fan, Z.; Liu, W.; Lei, Y.; Zhu, C.; Ma, B. Dihydrobenzoxazinone Derivatives as Aldose Reductase Inhibitors with Antioxidant Activity. Bioorg. Med. Chem. 2020, 28, 115699. [Google Scholar] [CrossRef]
- Fatmawati, S.; Ersam, T.; Yu, H.; Zhang, C.; Jin, F.; Shimizu, K. 20(S)-Ginsenoside Rh2 as Aldose Reductase Inhibitor from Panax ginseng. Bioorg. Med. Chem. Lett. 2014, 24, 4407–4409. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, M.T.; Imran, A.; Njateng, G.S.S.; Hameed, A.; Islam, M.; Al-Rashida, M.; Uroos, M.; Asari, A.; Shafiq, Z.; Iqbal, J. Benzoxazinone-Thiosemicarbazones as Antidiabetic Leads via Aldose Reductase Inhibition: Synthesis, Biological Screening and Molecular Docking Study. Bioorg. Chem. 2019, 87, 857–866. [Google Scholar] [CrossRef]
- Sorbinil Retinopathy Trial Research Group. A Randomized Trial of Sorbinil, an Aldose Reductase Inhibitor, in Diabetic Retinopathy. Arch. Ophthalmol. 1990, 108, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Sorbinil Retinopathy Trial Research Group. The Sorbinil Retinopathy Trial: Neuropathy Results. Neurology 1993, 43, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Christen, W.G.; Manson, J.E.; Bubes, V.; Glynn, R.J. Risk Factors for Progression of Distal Symmetric Polyneuropathy in Type 1 Diabetes mellitus. Am. J. Epidemiol. 1999, 150, 1142–1151. [Google Scholar] [CrossRef]
- Christensen, J.E.; Varnek, L.; Gregersen, G. The Effect of an Aldose Reductase Inhibitor (Sorbinil) on Diabetic Neuropathy and Neural Function of the Retina: A Double-Blind Study. Acta Neurol. Scand. 1985, 71, 164–167. [Google Scholar] [CrossRef]
- Cogan, D.G.; Kinoshita, J.H.; Kador, P.F.; Robison, G.; Datilis, M.B.; Cobo, L.M.; Kupfer, C. Aldose Reductase and Complications of Diabetes. Ann. Intern. Med. 1984, 101, 82–91. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.G.; Mota, C.C.; Leite, E.C.; Abreu, J.R.; Ruas, M.A. Effect of Sorbinil on Blood-Retinal Barrier in Early Diabetic Retinopathy. Diabetes 1986, 35, 574–578. [Google Scholar] [CrossRef]
- Grewal, A.S.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B.S. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-Diabetic Diseases. Mini Rev. Med. Chem. 2016, 16, 120–162. [Google Scholar] [CrossRef]
- Asano, T.; Saito, Y.; Kawakami, M.; Yamada, N. Fidarestat (SNK-860), A Potent Aldose Reductase Inhibitor, Normalizes the Elevated Sorbitol Accumulation in Erythrocytes of Diabetic Patients. J. Diabetes Complicat. 2002, 16, 133–138. [Google Scholar] [CrossRef]
- Hotta, N.; Toyota, T.; Matsuoka, K.; Shigeta, Y.; Kikkawa, R.; Kaneko, T.; Takahashi, A.; Sugimura, K.; Koike, Y.; Ishii, J.; et al. Clinical Efficacy of Fidarestat, a Novel Aldose Reductase Inhibitor, for Diabetic Peripheral Neuropathy: A 52-Week Multicenter Placebo-Controlled Double-Blind Parallel Group Study. Diabetes Care 2001, 24, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Chylack, L.T., Jr.; Henriques, H.F., 3rd; Cheng, H.M.; Tung, W.H. Efficacy of Alrestatin, an Aldose Reductase Inhibitor, in Human Diabetic and Nondiabetic Lenses. Ophthalmology 1979, 86, 1579–1585. [Google Scholar] [CrossRef]
- Distiller, L.A.; Joffe, B.I.; Sandler, M.; Kark, A.; Seftel, H.C. The Effect of Alrestatin on Alanine-Stimulated Release of Insulin and Glucagon in Man. J. Endocrinol. Investig. 1981, 4, 115–117. [Google Scholar] [CrossRef]
- Ehrig, T.; Bohren, K.M.; Prendergast, F.G.; Gabbay, K.H. Mechanism of Aldose Reductase Inhibition: Binding of NADP+/NADPH and Alrestatin-Like Inhibitors. Biochemistry 1994, 33, 7157–7165. [Google Scholar] [CrossRef] [PubMed]
- Gabbay, K.H.; Spack, N.; Loo, S.; Hirsch, H.J.; Ackil, A.A. Aldose Reductase Inhibition: Studies with Alrestatin. Metab. Clin. Exp. 1979, 28, 471–476. [Google Scholar] [CrossRef]
- Harrison, D.H.; Bohren, K.M.; Petsko, G.A.; Ringe, D.; Gabbay, K.H. The Alrestatin Double-Decker: Binding of Two Inhibitor Molecules to Human Aldose Reductase Reveals a New Specificity Deter-Minant. Biochemistry 1997, 36, 16134–16140. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, W.; Kobric, M. Effect of Alrestatin on Arginine-Induced Secretion of Glucagon and Insulin in the Rat. Horm. Metab. Res. 1978, 10, 280–282. [Google Scholar] [CrossRef]
- Maragoudakis, M.E.; Wasvary, J.; Hankin, H.; Gargiulo, P. Human Placenta Aldose Reductase. Forms Sensitive and Insensitive to Inhibition by Alrestatin. Mol. Pharmacol. 1984, 25, 425–430. [Google Scholar]
- O’Brien, M.M.; Schofield, P.J.; Edwards, M.R. Inhibition of Human Brain Aldose Reductase and Hexonate Dehydrogenase by Alrestatin and Sorbinil. J. Neurochem. 1982, 39, 810–814. [Google Scholar] [CrossRef]
- Tsai, S.C.; Burnakis, T.G. Aldose Reductase Inhibitors: An Update. Ann. Pharmacother. 1993, 27, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Cayen, M.N.; Hicks, D.R.; Ferdinandi, E.S.; Kraml, M.; Greselin, E.; Dvornik, D. Metabolic Disposition and Pharmacokinetics of the Aldose Reductase Inhibitor Tolrestat in Rats, Dogs, and Monkeys. Drug Metab. Dispos. Biol. Fate Chem. 1985, 13, 412–419. [Google Scholar] [PubMed]
- Daniele, E.; Coco, M.P. Tolrestat in the Therapy of Diabetic Peripheral Neuropathy: Is This Drug Really Useful? La Clin. Ter. 1995, 146, 793–799. [Google Scholar]
- Dvornik, D.; Millen, J.; Hicks, D.R.; Kraml, M. Tolrestat Pharmacokinetics in Rat Peripheral Nerve. J. Diabetes Complicat. 1994, 8, 18–26. [Google Scholar] [CrossRef]
- Fabiani, F.; De Vincentis, N.; Staffilano, A. Effect of Tolrestat on Oesophageal Transit Time and Cholecystic Motility in Type 2 Diabetic Patients with Asymptomatic Diabetic Neuropathy. Diabetes Metab. 1995, 21, 360–364. [Google Scholar]
- Giugliano, D.; Marfella, R.; Quatraro, A.; De Rosa, N.; Salvatore, T.; Cozzolino, D.; Ceriello, A.; Torella, R. Tolrestat for Mild Diabetic Neuropathy. A 52-Week, Randomized, Placebo-Controlled Trial. Ann. Intern. Med. 1993, 118, 7–11. [Google Scholar] [CrossRef]
- Hicks, D.R.; Kraml, M.; Cayen, M.N.; Dubuc, J.; Ryder, S.; Dvornik, D. Tolrestat Kinetics. Clin. Pharmacol. Ther. 1984, 36, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Rosenstock, J.; Challis, P.; Ryder, S.; Mullane, J.F.; Gonzalez, R.; Hicks, D.; Smith, T.; Dvornik, D. Effect of Tolrestat on Red Blood Cell Sorbitol Levels in Patients with Diabetes. Clin. Pharmacol. Ther. 1985, 38, 625–630. [Google Scholar] [CrossRef]
- Terada, M.; Yasuda, H.; Kikkawa, R.; Shigeta, Y. Tolrestat Improves Nerve Regeneration after Crush Injury in Streptozocin-Induced Diabetic Rats. Metab. Clin. Exp. 1996, 45, 1189–1195. [Google Scholar] [CrossRef]
- Van Griensven, J.M.; Jusko, W.J.; Lemkes, H.H.; Kroon, R.; Verhorst, C.J.; Chiang, S.T.; Cohen, A.F. Tolrestat Pharmacokinetic and Pharmacodynamic Effects on Red Blood Cell Sorbitol Levels in Normal Volunteers and in Patients with Insulin-Dependent Diabetes. Clin. Pharmacol. Ther. 1995, 58, 631–640. [Google Scholar] [CrossRef]
- Zhang, C.; Min, Z.; Liu, X.; Wang, C.; Wang, Z.; Shen, J.; Tang, W.; Zhang, X.; Liu, D.; Xu, X. Tolrestat Acts Atypically as a Competitive Inhibitor of the Thermostable Aldo-Keto Reductase Tm1743 from Thermotoga Mariti-Ma. FEBS Lett. 2020, 594, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rivera, F.; Concheiro, A.; Alvarez-Lorenzo, C. Epalrestat-Loaded Silicone Hydrogels as Contact Lenses to Address Diabetic-Eye Complications. Eur. J. Pharm. Biopharm. 2018, 122, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Geng, N.; Jin, Y.; Li, Y.; Zhu, S.; Bai, H. AKR1B10 Inhibitor Epalrestat Facilitates Sorafenib-Induced Apoptosis and Autophagy Via Targeting the mTOR Pathway in Hepatocellular Carcinoma. Int. J. Med. Sci. 2020, 17, 1246–1256. [Google Scholar] [CrossRef]
- He, J.; Gao, H.X.; Yang, N.; Zhu, X.D.; Sun, R.B.; Xie, Y.; Zeng, C.H.; Zhang, J.W.; Wang, J.K.; Ding, F.; et al. The Aldose Reductase Inhibitor Epalrestat Exerts Nephritic Protection on Diabetic Nephropathy in DB/DB Mice through Metabolic Modulation. Acta Pharmacol. Sin. 2019, 40, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Hotta, N. Epalrestat. Jpn. J. Clin. Med. 1997, 55, 204–211. [Google Scholar]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef]
- Steele, J.W.; Faulds, D.; Goa, K.L. Epalrestat. A Review of Its Pharmacology, and Therapeutic Potential in Late-Onset Complications of Diabetes mellitus. Drugs Aging 1993, 3, 532–555. [Google Scholar] [CrossRef]
- Tatsunami, R.; Murao, Y.; Sato, K. Protective Effect of Epalrestat against Oxidative Stress-induced Cytotoxicity. J. Pharm. Soc. Jpn. 2020, 140, 1381–1388. [Google Scholar] [CrossRef]
- Wang, X.; Lin, H.; Xu, S.; Jin, Y.; Zhang, R. Alpha Lipoic Acid Combined with Epalrestat: A Therapeutic Option for Patients with Diabetic Peripheral Neuropathy. Drug Des. Dev. Ther. 2018, 12, 2827–2840. [Google Scholar] [CrossRef]
- Yang, B.B.; Hong, Z.W.; Zhang, Z.; Yu, W.; Song, T.; Zhu, L.L.; Jiang, H.S.; Chen, G.T.; Chen, Y.; Dai, Y.T. Epalrestat, an Aldose Reductase Inhibitor, Restores Erectile Function in Streptozocin-Induced Diabetic Rats. Int. J. Impot. Res. 2019, 31, 97–104. [Google Scholar] [CrossRef]
- Boland, O.M.; Blackwell, C.C.; Clarke, B.F.; Ewing, D.J. Effects of Ponalrestat, an Aldose Reductase Inhibitor, on Neutrophil Killing of Escherichia coli and Autonomic Function in Patients with Diabetes mellitus. Diabetes 1993, 42, 336–340. [Google Scholar] [CrossRef]
- Kawamura, I.; Lacey, E.; Yamamoto, N.; Sakai, F.; Takeshita, S.; Inami, M.; Nishigaki, F.; Naoe, Y.; Tsujimoto, S.; Manda, T.; et al. Ponalrestat, an Aldose Reductase Inhibitor, Inhibits Cachexia Syndrome Induced by colon26 Adenocarcinoma in Mice. Anticancer Res. 1999, 19, 4105–4111. [Google Scholar] [PubMed]
- Kawamura, I.; Yamamoto, N.; Sakai, F.; Yamazaki, H.; Naoe, Y.; Inami, M.; Manda, T.; Shimomura, K. Activation of Lipoprotein Lipase and Inhibition of B16 Melanoma-Induced Cachexia in Mice by Ponalrestat, an Aldose Reductase Inhibitor. Anticancer Res. 1999, 19, 341–348. [Google Scholar] [PubMed]
- Moulds, R.F.; Fullinfaw, R.O.; Bury, R.W.; Plehwe, W.E.; Jacka, N.; McGrath, K.M.; Martin, F.I. Ponalrestat Does Not Cause a Protein Binding Interaction with Warfarin in Diabetic Patients. Br. J. Clin. Pharmacol. 1991, 31, 715–718. [Google Scholar] [CrossRef]
- Ramirez, L.C.; Arauz, C.; Pruneda, L.; Hammon, K.; Rosenstock, J.; Raskin, P. The Effect of Aldose Reductase Inhibition with Ponalrestat on the Width of the Capillary Basement Membrane in Diabetes mellitus. Diabetes Res. Clin. Pract. 1991, 11, 73–80. [Google Scholar] [CrossRef]
- Sima, A.A.; Prashar, A.; Zhang, W.X.; Chakrabarti, S.; Greene, D.A. Preventive Effect of Long-Term Aldose Reductase Inhibition (Ponalrestat) on Nerve Conduction and Sural Nerve Structure in the Spontaneously Diabetic Bio-Breeding Rat. J. Clin. Investig. 1990, 85, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Sundkvist, G.; Armstrong, F.M.; Bradbury, J.E.; Chaplin, C.; Ellis, S.H.; Owens, D.R.; Rosén, I.; Sönksen, P. Peripheral and Autonomic Nerve Function in 259 Diabetic Patients with Peripheral Neuropathy Treated with Ponalrestat (an Aldose Reductase Inhibitor) or Placebo for 18 Months. J. Diabetes Complicat. 1992, 6, 123–130. [Google Scholar] [CrossRef]
- Ward, W.H.; Sennitt, C.M.; Ross, H.; Dingle, A.; Timms, D.; Mirrlees, D.J.; Tuffin, D.P. Ponalrestat: A Potent and Specific Inhibitor of Aldose Reductase. Biochem. Pharmacol. 1990, 39, 337–346. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, H.J.; Su, Q.; Wen, J.; Wang, Y.; Song, W.; Xie, Y.; He, W.; Yang, Z.; Jiang, K.; et al. A Phenotype-Directed Chemical Screen Identifies Ponalrestat as an Inhibitor of the Plant Flavin Monooxygenase YUCCA in Auxin Biosynthesis. J. Biol. Chem. 2019, 294, 19923–19933. [Google Scholar] [CrossRef]
- Ziegler, D.; Mayer, P.; Rathmann, W.; Gries, F.A. One-Year Treatment with the Aldose Reductase Inhibitor, Ponalrestat, in Diabetic Neuropathy. Diabetes Res. Clin. Pract. 1991, 14, 63–73. [Google Scholar] [CrossRef]
- Abdel Motaal, A.; El-Askary, H.; Crockett, S.; Kunert, O.; Sakr, B.; Shaker, S.; Grigore, A.; Albulescu, R.; Bauer, R. Aldose Reductase Inhibition of a Saponin-Rich Fraction and New Furostanol saponin Derivatives from Balanites aegyptiaca. Phytomed. Int. J. Phytother. Phytopharm. 2015, 22, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Bhadada, S.V.; Vyas, V.K.; Goyal, R.K. Protective Effect of Tephrosia purpurea in Diabetic Cataract through Aldose Reductase Inhibitory Activity. Biomed. Pharmacother. 2016, 83, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Head, K.A. Natural Therapies for Ocular Disorders, Part Two: Cataracts and Glaucoma. Altern. Med. Rev. A J. Clin. Ther. 2001, 6, 141–166. [Google Scholar]
- Huang, G.J.; Hsieh, W.T.; Chang, H.Y.; Huang, S.S.; Lin, Y.C.; Kuo, Y.H. α-Glucosidase and Aldose Reductase Inhibitory Activities from the Fruiting Body of Phellinus merrillii. J. Agric. Food Chem. 2011, 59, 5702–5706. [Google Scholar] [CrossRef] [PubMed]
- Kador, P.F.; O’Meara, J.D.; Blessing, K.; Marx, D.B.; Reinhardt, R.A. Efficacy of Structurally Diverse Aldose Reductase Inhibitors on Experimental Periodontitis in Rats. J. Periodontol. 2011, 82, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Milackova, I.; Prnova, M.S.; Majekova, M.; Sotnikova, R.; Stasko, M.; Kovacikova, L.; Banerjee, S.; Veverka, M.; Stefek, M. 2-Chloro-1,4-Naphthoquinone Derivative of Quercetin as an Inhibitor of Aldose Reductase and Anti-Inflammatory Agent. J. Enzym. Inhib. Med. Chem. 2015, 30, 107–113. [Google Scholar] [CrossRef]
- Morjana, N.A.; Flynn, T.G. Aldose Reductase from Human Psoas Muscle: Purification, Substrate Specificity, Immunological Characterization, and Effect of Drugs and Inhibitors. J. Biol. Chem. 1989, 264, 2906–2911. [Google Scholar] [CrossRef]
- Sever, B.; Altıntop, M.D.; Demir, Y.; Akalın Çiftçi, G.; Beydemir, Ş.; Özdemir, A. Design, Synthesis, In Vitro and In Silico Investigation of Aldose Reductase Inhibitory Effects of New Thiazole-Based Compounds. Bioorg. Chem. 2020, 102, 104110. [Google Scholar] [CrossRef]
- Bril, V.; Buchanan, R.A. Aldose Reductase Inhibition by AS-3201 in Sural Nerve From Patients with Diabetic Sensorimotor Polyneuropathy. Diabetes Care 2004, 27, 2369–2375. [Google Scholar] [CrossRef][Green Version]
- Chalk, C.; Benstead, T.J.; Moore, F. Aldose Reductase Inhibitors for the Treatment of Diabetic Polyneuropathy. Cochrane Database Syst. Rev. 2007, 2007, Cd004572. [Google Scholar] [CrossRef]
- Giannoukakis, N. Ranirestat as a Therapeutic Aldose Reductase Inhibitor for Diabetic Complications. Expert Opin. Investig. Drugs 2008, 17, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kurono, M.; Fujiwara, I.; Yoshida, K. Stereospecific Interaction of a Novel Spirosuccinimide Type Aldose Reductase Inhibitor, AS-3201, with Aldose Reductase. Biochemistry 2001, 40, 8216–8226. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ono, Y.; Kuromiya, A.; Toyosawa, K.; Ueda, Y.; Bril, V. Long-Term Treatment with Ranirestat (AS-3201), a Potent Aldose Reductase Inhibitor, Suppresses Diabetic Neuropathy and Cata-Ract Formation in Rats. J. Pharmacol. Sci. 2008, 107, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ono, Y.; Kurono, M.; Kuromiya, A.; Nakamura, K.; Bril, V. Ranirestat (AS-3201), a Potent Aldose Reductase Inhibitor, Reduces Sorbitol Levels and Improves Motor Nerve Conduction Velocity in Streptozotocin-Diabetic Rats. J. Pharmacol. Sci. 2008, 107, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Negoro, T.; Murata, M.; Ueda, S.; Fujitani, B.; Ono, Y.; Kuromiya, A.; Komiya, M.; Suzuki, K.; Matsumoto, J. Novel, Highly Potent Aldose Reductase Inhibitors: (R)-(-)-2-(4-Bromo-2-Fluorobenzyl)-1,2, 3,4- tetrahydropyrrolo[1,2-A]pyrazine -4-Spiro-3′-Pyrrolidine-1,2′, 3,5′-Tetrone (AS-3201) and Its Congeners. J. Med. Chem. 1998, 41, 4118–4129. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Kohara, N.; Baba, M.; Komori, T.; Naito, Y.; Imai, T.; Satoh, J.; Yamaguchi, Y.; Hamatani, T. Aldose Reductase Inhibitor Ranirestat Significantly Improves Nerve Conduction Velocity in Diabetic Polyneuropathy: A Randomized Double-Blind Placebo-Controlled Study in Japan. J. Diabetes Investig. 2019, 10, 466–474. [Google Scholar] [CrossRef]
- Ishii, A.; Kotani, T.; Nagaki, Y.; Shibayama, Y.; Toyomaki, Y.; Okukado, N.; Ienaga, K.; Okamoto, K. Highly Selective Aldose Reductase Inhibitors. 1. 3-(Arylalkyl)-2,4, 5-Trioxoimidazolidine-1-Acetic Acids. J. Med. Chem. 1996, 39, 1924–1927. [Google Scholar] [CrossRef]
- Kotani, T.; Nagaki, Y.; Ishii, A.; Konishi, Y.; Yago, H.; Suehiro, S.; Okukado, N.; Okamoto, K. Highly Selective Aldose Reductase Inhibitors. 3. Structural Diversity of 3-(Arylmethyl)-2,4, 5-Trioxoimidazolidine-1-Acetic Acids. J. Med. Chem. 1997, 40, 684–694. [Google Scholar] [CrossRef]
- Nakamura, J.; Hamada, Y.; Chaya, S.; Nakashima, E.; Naruse, K.; Kato, K.; Yasuda, Y.; Kamiya, H.; Sakakibara, F.; Koh, N.; et al. Transition Metals and Polyol Pathway in the Development of Diabetic Neuropathy in Rats. Diabetes Metab. Res. Rev. 2002, 18, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Kato, K.; Hamada, Y.; Nakayama, M.; Chaya, S.; Nakashima, E.; Naruse, K.; Kasuya, Y.; Mizubayashi, R.; Miwa, K.; et al. A Protein Kinase C-Beta-Selective Inhibitor Ameliorates Neural Dysfunction in Streptozotocin-Induced Diabetic Rats. Diabetes 1999, 48, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, V.F.; Barreto, E.O.; Serra, M.F.; Cordeiro, R.S.; Martins, M.A.; Fortes, Z.B.; e Silva, P.M. Aldose Reductase Inhibitor Zopolrestat Restores Allergic Hyporesponsiveness in Alloxan-Diabetic Rats. Eur. J. Pharmacol. 2006, 549, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Inskeep, P.B.; Reed, A.E.; Ronfeld, R.A. Pharmacokinetics of Zopolrestat, a Carboxylic Acid Aldose Reductase Inhibitor, in Normal and Diabetic Rats. Pharm. Res. 1991, 8, 1511–1515. [Google Scholar] [CrossRef]
- Inskeep, P.B.; Ronfeld, R.A.; Peterson, M.J.; Gerber, N. Pharmacokinetics of the Aldose Reductase Inhibitor, Zopolrestat, in Humans. J. Clin. Pharmacol. 1994, 34, 760–766. [Google Scholar] [CrossRef]
- Mylari, B.L.; Larson, E.R.; Beyer, T.A.; Zembrowski, W.J.; Aldinger, C.E.; Dee, M.F.; Siegel, T.W.; Singleton, D.H. Novel, Potent Aldose Reductase Inhibitors: 3,4-Dihydro-4-Oxo-3-[[5-(Trifluoromethyl)-2-Benzothiazolyl] Methyl]-1-Phthalazineacetic Acid (Zopolrestat) and Congeners. J. Med. Chem. 1991, 34, 108–122. [Google Scholar] [CrossRef]
- Qiu, L.; Cai, C.; Zhao, X.; Fang, Y.; Tang, W.; Guo, C. Inhibition of Aldose Reductase Ameliorates Ethanol-Induced Steatosis in HepG2 Cells. Mol. Med. Rep. 2017, 15, 2732–2736. [Google Scholar] [CrossRef]
- Ramasamy, R.; Liu, H.; Oates, P.J.; Schaefer, S. Attenuation of Ischemia Induced Increases in Sodium and Calcium by the Aldose Reductase Inhibitor Zopolrestat. Cardiovasc. Res. 1999, 42, 130–139. [Google Scholar] [CrossRef]
- Schneider, R.P.; Davenport, C.J.; Hoffmaster, K.A.; Inskeep, P.B. Bioavailability, Multiple-Dose Pharmacokinetics, and Biotransformation of the Aldose Reductase Inhibitor Zopolrestat in Dogs. Drug Metab. Dispos. Biol. Fate Chem. 1998, 26, 1160–1166. [Google Scholar] [PubMed]
- Schneider, R.P.; Fouda, H.G.; Inskeep, P.B. Tissue Distribution and Biotransformation of Zopolrestat, an Aldose Reductase Inhibitor, in Rats. Drug Metab. Dispos. Biol. Fate Chem. 1998, 26, 1149–1159. [Google Scholar]
- Tracey, W.R.; Magee, W.P.; Ellery, C.A.; MacAndrew, J.T.; Smith, A.H.; Knight, D.R.; Oates, P.J. Aldose Reductase Inhibition Alone or Combined with an Adenosine A(3) Agonist Reduces Ischemic Myocardial Injury. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 1447–1452. [Google Scholar] [CrossRef]
- Florkowski, C.M.; Rowe, B.R.; Nightingale, S.; Harvey, T.C.; Barnett, A.H. Clinical and Neurophysiological Studies of Aldose Reductase Inhibitor Ponalrestat in Chronic Symptomatic Diabetic Peripheral Neuropathy. Diabetes 1991, 40, 129–133. [Google Scholar] [CrossRef]
- Wang, Z.L.; Deng, C.Y.; Zheng, H.; Xie, C.F.; Wang, X.H.; Luo, Y.F.; Chen, Z.Z.; Cheng, P.; Chen, L.J. (Z)2-(5-(4-Methoxybenzylidene)-2, 4-Dioxothiazolidin-3-Yl) Acetic Acid Protects Rats from CCl(4)-Induced Liver Injury. J. Gastroenterol. Hepatol. 2012, 27, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.A.; Arezzo, J.C.; Brown, M.B. Effect of Aldose Reductase Inhibition on Nerve Conduction and Morphometry in Diabetic Neuropathy. Neurology 1999, 53, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Miyake, H.; Fujii, T.; Takakura, S.; Goto, T. The Structure of Human Recombinant Aldose Reductase Complexed with the Potent Inhibitor Zenarestat. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 622–626. [Google Scholar] [CrossRef]
- Oka, M.; Kato, N. Aldose Reductase Inhibitors. J. Enzym. Inhib. 2001, 16, 465–473. [Google Scholar] [CrossRef]
- Shimoshige, Y.; Ikuma, K.; Yamamoto, T.; Takakura, S.; Kawamura, I.; Seki, J.; Mutoh, S.; Goto, T. The Effects of Zenarestat, an Aldose Reductase Inhibitor, on Peripheral Neuropathy in Zucker Diabetic Fatty Rats. Metab. Clin. Exp. 2000, 49, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Shimoshige, Y.; Minoura, K.; Matsuoka, N.; Takakura, S.; Mutoh, S.; Kamijo, M. Thirteen-Month Inhibition of Aldose Reductase by Zenarestat Prevents Morphological Abnormalities in the Dorsal Root Ganglia of Streptozotocin-Induced Diabetic Rats. Brain Res. 2009, 1247, 182–187. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sawamoto, T.; Suzuki, A.; Kimura, T. Pharmacokinetics of Zenarestat, an Aldose Reductase Inhibitor, in Male and Female Diabetic Rats. Drug Metab. Dispos. Biol. Fate Chem. 1993, 21, 677–681. [Google Scholar]
- Tanaka, Y.; Sekiguchi, M.; Sawamoto, T.; Katami, Y.; Ueda, T.; Esumi, Y.; Noda, K. Absorption, Distribution and Excretion of Zenarestat, a New Aldose Reductase Inhibitor, in Rats and Dogs. Xenobiotica Fate Foreign Compd. Biol. Syst. 1992, 22, 57–64. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimojyo, H.; Hata, T.; Hashimoto, M.; Noguchi, H. Toxicokinetics of Zenarestat, an Aldose Reductase Inhibitor in Animals and Man. Xenobiotica Fate Foreign Compd. Biol. Syst. 1994, 24, 461–471. [Google Scholar] [CrossRef]
- Pastel, E.; Pointud, J.C.; Volat, F.; Martinez, A.; Lefrancois-Martinez, A.M. Aldo-Keto Reductases 1B in Endocrinology and Metabolism. Front. Pharmacol. 2012, 3, 148. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Moro, A.; Gallego, O.; Ardevol, A.; Rovira, C.; Petrash, J.M.; Pares, X.; Farres, J. Human and Rodent Aldo-Keto Reductases from the AKR1B Subfamily and Their Specificity with Retinaldehyde. Chem. Biol. Interact. 2011, 191, 199–205. [Google Scholar] [CrossRef]
- Danesh, F.R.; Wada, J.; Wallner, E.I.; Sahai, A.; Srivastava, S.K.; Kanwar, Y.S. Gene Regulation of Aldose-, Aldehyde- and a Renal Specific Oxido Reductase (RSOR) in the Pathobiology of Diabetes mellitus. Curr. Med. Chem. 2003, 10, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Dixit, B.L.; Balendiran, G.K.; Watowich, S.J.; Srivastava, S.; Ramana, K.V.; Petrash, J.M.; Bhatnagar, A.; Srivastava, S.K. Kinetic and Structural Characterization of the Glutathione-Binding Site of Aldose Reductase. J. Biol. Chem. 2000, 275, 21587–21595. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Chandra, A.; Bhatnagar, A.; Srivastava, S.K.; Ansari, N.H. Lipid Peroxidation Product, 4-Hydroxynonenal and Its Conjugate with GSH are Excellent Substrates of Bovine Lens Aldose Reductase. Biochem. Biophys. Res. Commun. 1995, 217, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Chung, S.S.; Kador, P.F. Diabetic Cataracts: Mechanisms and Management. Diabetes Metab. Res. Rev. 2010, 26, 172–180. [Google Scholar] [CrossRef]
- Lorenzi, M. The Polyol Pathway as a Mechanism for Diabetic Retinopathy: Attractive, Elusive, and Resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Oates, P.J. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Curr. Drug Targets 2008, 9, 14–36. [Google Scholar] [CrossRef]
- Dunlop, M. Aldose Reductase and the Role of the Polyol Pathway in Diabetic Nephropathy. Kidney Int. 2000, 77, 3–12. [Google Scholar] [CrossRef]
- Chung, S.S.; Chung, S.K. Aldose Reductase in Diabetic Microvascular Complications. Curr. Drug Targets 2005, 6, 475–486. [Google Scholar] [CrossRef]
- Li, Q.; Hwang, Y.C.; Ananthakrishnan, R.; Oates, P.J.; Guberski, D.; Ramasamy, R. Polyol Pathway and Modulation of Ischemia-Reperfusion Injury in Type 2 Diabetic BBZ Rat Hearts. Cardiovasc. Diabetol. 2008, 7, 33. [Google Scholar] [CrossRef]
- Srivastava, S.K.; Ramana, K.V.; Bhatnagar, A. Role of Aldose Reductase and Oxidative Damage in Diabetes and the Consequent Potential for Therapeutic Options. Endocr. Rev. 2005, 26, 380–392. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chung, S.K.; Chung, S.S. Demonstration that Polyol Accumulation is Responsible for Diabetic Cataract by the Use of Transgenic Mice Expressing the Al-Dose Reductase Gene in the Lens. Proc. Natl. Acad. Sci. USA 1995, 92, 2780–2784. [Google Scholar] [CrossRef]
- Suzen, S.; Buyukbingol, E. Recent Studies of Aldose Reductase Enzyme Inhibition for Diabetic Complications. Curr. Med. Chem. 2003, 10, 1329–1352. [Google Scholar] [CrossRef]
- Ito, A.; Ishii-Nozawa, R.; Ibuki, C.; Atarashi, H.; Kataoka, H.; Takeuchi, K. Examination of Questionnaires Regarding Diabetic Peripheral Neuropathy in Epalrestat-Treated Patients and Their Usefulness in the Treatment of the Patients during the Treatment Course. J. Pharm. Soc. Jpn. 2009, 129, 1239–1247. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kubo, E.; Mori, K.; Kobayashi, T.; Takahashi, Y.; Yokoi, N.; Kinoshita, S.; Kasahara, T.; Yonezawa, H.; Akagi, Y. Effect of Aldose Reductase Inhibitor on Corneal Epithelial Barrier Function in Galactose-Fed Dogs. J. Ocul. Pharmacol. Ther. 1998, 14, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.H.; Bhatnagar, A.; Fulep, E.; Khanna, P.; Srivastava, S.K. Trolox Protects Hyperglycemia-Induced Cataractogenesis in Cultured Rat Lens. Res. Commun. Chem. Pathol. Pharmacol. 1994, 84, 93–104. [Google Scholar] [PubMed]
- Srivastava, S.K.; Ansari, N.H. Prevention of Sugar-Induced Cataractogenesis in Rats by Butylated Hydroxytoluene. Diabetes 1988, 37, 1505–1508. [Google Scholar] [CrossRef]
- Ramana, K.V.; Bhatnagar, A.; Srivastava, S.; Yadav, U.C.; Awasthi, S.; Awasthi, Y.C.; Srivastava, S.K. Mitogenic Responses of Vascular Smooth Muscle Cells to Lipid Peroxidation-Derived Aldehyde 4-Hydroxy-Trans-2-Nonenal (HNE): Role of Aldose Reductase-Catalyzed Reduction of the HNE-Glutathione Conjugates in Regulating Cell Growth. J. Biol. Chem. 2006, 281, 17652–17660. [Google Scholar] [CrossRef]
- Ramana, K.V.; Chandra, D.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Aldose Reductase Mediates the Mitogenic Signals of Cytokines. Chem. Biol. Interact. 2003, 143, 587–596. [Google Scholar] [CrossRef]
- Chandra, D.; Ramana, K.V.; Friedrich, B.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Role of Aldose Reductase in TNF-Alpha-Induced Apoptosis of Vascular Endothelial Cells. Chem. Biol. Interact. 2003, 143, 605–612. [Google Scholar] [CrossRef]
- Shinmura, K.; Bolli, R.; Liu, S.Q.; Tang, X.L.; Kodani, E.; Xuan, Y.T.; Srivastava, S.; Bhatnagar, A. Aldose Reductase Is an Obligatory Mediator of the Late Phase of Ischemic Preconditioning. Circ. Res. 2002, 91, 240–246. [Google Scholar] [CrossRef]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose Reductase, Oxidative Stress, and Diabetic Mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Gupta, S.K.; Ali, V.; Singh, P.; Verma, M. Aldose Reductase: A Cause and a Potential Target for the Treatment of Diabetic Complications. Arch. Pharmacal Res. 2021, 44, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V. Aldose Reductase: New Insights for an Old Enzyme. Biomol. Concepts 2011, 2, 103–114. [Google Scholar] [CrossRef]
- Srivastava, S.; Ramana, K.V.; Tammali, R.; Srivastava, S.K.; Bhatnagar, A. Contribution of Aldose Reductase to Diabetic Hyperproliferation of Vascular Smooth Muscle Cells. Diabetes 2006, 55, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Reddy, A.B.; Srivastava, S.K.; Ramana, K.V. Inhibition of Aldose Reductase Prevents Angiogenesis In Vitro and In Vivo. Angiogenesis 2011, 14, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Fu, J.; Tay, S.S.; Ling, E.A.; Dheen, S.T. Aldose Reductase is Implicated in High Glucose-Induced Oxidative Stress in Mouse Embryonic Neural Stem Cells. J. Neurochem. 2007, 103, 1654–1665. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; Sanders, J.M.; Nadler, J.; Ley, K. Upregulation of Aldose Reductase during Foam Cell Formation as Possible Link among Diabetes, Hyperlipidemia, and Atheroscle-Rosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1137–1143. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose Reductase-Mediated Phosphorylation of p53 Leads to Mitochondrial Dysfunction and Damage in Diabetic Platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- King, G.L.; Shiba, T.; Oliver, J.; Inoguchi, T.; Bursell, S.E. Cellular and Molecular Abnormalities in the Vascular Endothelium of Diabetes mellitus. Annu. Rev. Med. 1994, 45, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, P.R.; Chen, H.Q.; Yang, J.; Yorio, T. Modulation of Myo-[3H] inositol Uptake by Glucose and Sorbitol in Cultured Bovine Lens Epithelial Cells. II. Characterization of High-And Low-Affinity Myo-Inositol Transport Sites. Investig. Ophthalmol. Vis. Sci. 1992, 33, 3572–3580. [Google Scholar]
- Kang, E.S.; Kim, H.J.; Paek, K.S.; Jang, H.S.; Chang, K.C.; Lee, J.H.; Nishinaka, T.; Yabe-Nishimura, C.; Seo, H.G. Phorbol Ester Up-Regulates Aldose Reductase Expression in A549 Cells: A Potential Role for Aldose Reductase in Cell Cycle Modu-Lation. Cell. Mol. Life Sci. 2005, 62, 1146–1155. [Google Scholar] [CrossRef]
- Yamaoka, T.; Oda, A.; Bannai, C.; Itakura, M.; Yamashita, K. The Effect of Non-Enzymatic Glycation on Recombinant Human Aldose Reductase. Diabetes Res. Clin. Pract. 1995, 27, 165–169. [Google Scholar] [CrossRef]
- Dan, Q.; Wong, R.; Chung, S.K.; Chung, S.S.; Lam, K.S. Interaction between the Polyol Pathway and Non-enzymatic Glycation on Aortic Smooth Muscle Cell Migration and Monocyte Adhesion. Life Sci. 2004, 76, 445–459. [Google Scholar] [CrossRef]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid Formation of Advanced Glycation End Products by Intermediate Metabolites of Glycolytic Pathway and Polyol Pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef]
- Nagaraj, R.H.; Prabhakaram, M.; Ortwerth, B.J.; Monnier, V.M. Suppression of Pentosidine Formation in Galactosemic Rat Lens by an Inhibitor of Aldose Reductase. Diabetes 1994, 43, 580–586. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, J.K.; Kang, Y.H.; Lee, J.Y.; Kang, I.J.; Lim, S.S. Aldose Reductase Inhibitory Activity of Compounds from Zea mays L. Biomed. Res. Int. 2013, 2013, 727143. [Google Scholar] [CrossRef]
- Ramana, K.V.; Friedrich, B.; Tammali, R.; West, M.B.; Bhatnagar, A.; Srivastava, S.K. Requirement of Aldose Reductase for the Hyperglycemic Activation of Protein Kinase C and Formation of Diacylglycerol in Vascular Smooth Muscle Cells. Diabetes 2005, 54, 818–829. [Google Scholar] [CrossRef]
- Ramana, K.V.; Chandra, D.; Srivastava, S.; Bhatnagar, A.; Aggarwal, B.B.; Srivastava, S.K. Aldose Reductase Mediates Mitogenic Signaling in Vascular Smooth Muscle Cells. J. Biol. Chem. 2002, 277, 32063–32070. [Google Scholar] [CrossRef]
- Ruef, J.; Liu, S.Q.; Bode, C.; Tocchi, M.; Srivastava, S.; Runge, M.S.; Bhatnagar, A. Involvement of Aldose Reductase in Vascular Smooth Muscle Cell Growth and Lesion Formation After Arterial Injury. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1745–1752. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Kaneko, M.; Bakr, S.; Liao, H.; Lu, Y.; Lewis, E.R.; Yan, S.; Ii, S.; Itakura, M.; Rui, L.; et al. Central Role for Aldose Reductase Pathway in Myocardial Ischemic Injury. FASEB J. 2004, 18, 1192–1199. [Google Scholar] [CrossRef]
- Srivastava, S.; Vladykovskaya, E.; Barski, O.A.; Spite, M.; Kaiserova, K.; Petrash, J.M.; Chung, S.S.; Hunt, G.; Dawn, B.; Bhatnagar, A. Aldose Reductase Protects against Early Atherosclerotic Lesion Formation in Apolipoprotein E-null Mice. Circ. Res. 2009, 105, 793–802. [Google Scholar] [CrossRef]
- Jannapureddy, S.; Sharma, M.; Yepuri, G.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase: An Emerging Target for Development of Interventions for Diabetic Cardiovascular Complications. Front. Endocrinol. 2021, 12, 636267. [Google Scholar] [CrossRef]
- Mizukami, H.; Osonoi, S. Pathogenesis and Molecular Treatment Strategies of Diabetic Neuropathy Collateral Glucose-Utilizing Pathways in Diabetic Polyneuropathy. Int. J. Mol. Sci. 2020, 22, 94. [Google Scholar] [CrossRef]
- Ramana, K.V.; Friedrich, B.; Bhatnagar, A.; Srivastava, S.K. Aldose Reductase Mediates Cytotoxic Signals of Hyperglycemia and TNF-Alpha in Human Lens Epithelial Cells. FASEB J. 2003, 17, 315–317. [Google Scholar] [CrossRef]
- Stevens, M.J.; Dananberg, J.; Feldman, E.L.; Lattimer, S.A.; Kamijo, M.; Thomas, T.P.; Shindo, H.; Sima, A.A.; Greene, D.A. The Linked Roles of Nitric Oxide, Aldose Reductase and, (Na+, K+)-ATPase in the Slowing of Nerve Conduction in the Strepto-Zotocin Diabetic Rat. J. Clin. Investig. 1994, 94, 853–859. [Google Scholar] [CrossRef]
- Suzuki, H.; Shimosegawa, T.; Ohara, S.; Toyota, T. Epalrestat Prevents the Decrease in Gastric Mucosal Blood Flow and Protects the Gastric Mucosa in Streptozotocin Diabetic Rats. J. Gastroenterol. 1999, 34, 172–177. [Google Scholar] [CrossRef]
- Kern, T.; Engerman, R. Development of Complications in Diabetic Dogs and Galactosemic Dogs: Effect of Aldose Reductase Inhibitors. In Proceedings of the Workshop on Aldose Reductase Inhibitors; NIH Publication: Bethesda, MD, USA, 1991; pp. 81–3114. [Google Scholar]
- Engerman, R.L.; Kern, T.S.; Garment, M.B. Capillary Basement Membrane in Retina, Kidney, and Muscle of Diabetic Dogs and Galactosemic Dogs and Its Response to 5 Years Aldose Reductase Inhibition. J. Diabetes Complicat. 1993, 7, 241–245. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and Collagen Regulate Human Platelet Activity through Aldose Reductase Induction of Thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.C.; Ramana, K.V.; Srivastava, S.K. Aldose Reductase Inhibition Suppresses Airway Inflammation. Chem. Biol. Interact. 2011, 191, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.C.; Ramana, K.V.; Aguilera-Aguirre, L.; Boldogh, I.; Boulares, H.A.; Srivastava, S.K. Inhibition of Aldose Reductase Prevents Experimental Allergic Airway Inflammation in Mice. PLoS ONE 2009, 4, e6535. [Google Scholar] [CrossRef] [PubMed]
- Yadav, U.C.; Naura, A.S.; Aguilera-Aguirre, L.; Boldogh, I.; Boulares, H.A.; Calhoun, W.J.; Ramana, K.V.; Srivastava, S.K. Aldose Reductase Inhibition Prevents Allergic Airway Remodeling through PI3K/AKT/GSK3 Beta Pathway in Mice. PLoS ONE 2013, 8, e57442. [Google Scholar] [CrossRef]
- Yadav, U.C.; Naura, A.S.; Aguilera-Aguirre, L.; Ramana, K.V.; Boldogh, I.; Sur, S.; Boulares, H.A.; Srivastava, S.K. Aldose Reductase Inhibition Suppresses the Expression of Th2 Cytokines and Airway Inflammation in Ovalbumin-Induced Asthma in Mice. J. Immunol. 2009, 183, 4723–4732. [Google Scholar] [CrossRef]
- Modjtahedi, B.S.; Wu, J.; Luong, T.Q.; Gandhi, N.K.; Fong, D.S.; Chen, W. Severity of Diabetic Retinopathy and the Risk of Future Cerebrovascular Disease, Cardiovascular Disease, and All-Cause Mortality. Ophthalmology 2021, 128, 1169–1179. [Google Scholar] [CrossRef]
- Roy, S.; Lorenzi, M. Early Biosynthetic Changes in the Diabetic-Like Retinopathy of Galactose-Fed Rats. Diabetologia 1996, 39, 735–738. [Google Scholar] [CrossRef]
- Segawa, M.; Hirata, Y.; Fujimori, S.; Okada, K. The Development of Electroretinogram Abnormalities and the Possible Role of Polyol Pathway Activity in Diabetic Hyperglyce-Mia and Galactosemia. Metab. Clin. Exp. 1988, 37, 454–460. [Google Scholar] [CrossRef]
- Sun, W.; Oates, P.J.; Coutcher, J.B.; Gerhardinger, C.; Lorenzi, M. A Selective Aldose Reductase Inhibitor of a New Structural Class Prevents or Reverses Early Retinal Abnormalities in Experimental Diabetic Retinopathy. Diabetes 2006, 55, 2757–2762. [Google Scholar] [CrossRef]
- Homme, R.P.; Sandhu, H.S.; George, A.K.; Tyagi, S.C.; Singh, M. Sustained Inhibition of NF-κB Activity Mitigates Retinal Vasculopathy in Diabetes. Am. J. Pathol. 2021, 191, 947–964. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Kador, P.F. Aldose Reductase/Polyol Inhibitors for Diabetic Retinopathy. Curr. Pharm. Biotechnol. 2011, 12, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.J.; Li, S.Y.; Kociok, N.; Wong, D.; Chung, S.K.; Lo, A.C. Aldose Reductase Deficiency Reduced Vascular Changes in Neonatal Mouse Retina in Oxygen-Induced Retinopathy. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5698–5712. [Google Scholar] [CrossRef][Green Version]
- Drel, V.R.; Pacher, P.; Ali, T.K.; Shin, J.; Julius, U.; El-Remessy, A.B.; Obrosova, I.G. Aldose Reductase Inhibitor Fidarestat Counteracts Diabetes-Associated Cataract Formation, Retinal Oxidative-Nitrosative Stress, Glial Activation, and Apoptosis. Int. J. Mol. Med. 2008, 21, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.; Shieh, B.; Chang, K.C.; Pal, A.; Lenhart, P.; Ammar, D.; Ruzycki, P.; Palla, S.; Reddy, G.B.; Petrash, J.M. Aldose Reductase Expression as a Risk Factor for Cataract. Chem. Biol. Interact. 2014, 234. [Google Scholar] [CrossRef] [PubMed]
- Tromp, A.; Hooymans, J.M.; Barendsen, B.C.; van Doormaal, J.J. The Effects of an Aldose Reductase Inhibitor on the Progression of Diabetic Retinopathy. Doc. Ophthalmol. 1991, 78, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, M.; Miyata, K.; Otani, S.; Miyai, T.; Nejima, R.; Yamagami, S.; Amano, S. A Randomised, Placebo Controlled Clinical Trial of the Aldose Reductase Inhibitor CT-112 as Management of Corneal Epithelial Disorders in Diabetic Patients. Br. J. Ophthalmol. 2005, 89, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, M.R.; Amiri, A.A.; Hashemi-Soteh, M.B.; Daneshvar, F.; Emady-Jamaly, R.; Jafari, R.; Soleimani, B.; Haghiaminjan, H. Aldose Reductase C-106T Gene Polymorphism in Type 2 Diabetics with Microangiopathy in Iranian Individuals. Indian J. Endocrinol. Metab. 2015, 19, 95–99. [Google Scholar] [CrossRef]
- Yadav, U.C.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Inhibition Prevents Endotoxin-Induced Uveitis in Rats. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4634–4642. [Google Scholar] [CrossRef]
- Ramana, K.V.; Srivastava, S.K. Aldose Reductase: A Novel Therapeutic Target for Inflammatory Pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20. [Google Scholar] [CrossRef]
- Yadav, U.C.; Shoeb, M.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Deficiency Protects from Autoimmune–And Endotoxin-Induced Uveitis in Mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8076–8085. [Google Scholar] [CrossRef]
- Sourris, K.C.; Forbes, J.M. Interactions between Advanced Glycation End-Products (AGE) and Their Receptors in the Development and Progression of Diabetic Nephropathy-Are These Receptors Valid Therapeutic Targets. Curr. Drug Targets 2009, 10, 42–50. [Google Scholar] [CrossRef]
- Ishii, H.; Tada, H.; Isogai, S. An Aldose Reductase Inhibitor Prevents Glucose-Induced Increase in Transforming Growth Factor-β and Protein Kinase C Activi-Ty in Cultured Human Mesangial Cells. Diabetologia 1998, 41, 362–364. [Google Scholar] [CrossRef][Green Version]
- Dan, Q.; Wong, R.L.; Yin, S.; Chung, S.K.; Chung, S.S.; Lam, K.S. Interaction between the Polyol Pathway and Non-Enzymatic Glycation on Mesangial Cell Gene Expression. Nephron Exp. Nephrol. 2004, 98, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Fujii, Y.; Kobashi, K.; Nomura, K. Aldose Reductase Inhibitors: Flavonoids, Alkaloids, Acetophenones, Benzophenones, and Spirohydantoins of Chroman. Arch. Biochem. Biophys. 1985, 239, 491–496. [Google Scholar] [CrossRef]
- Forbes, J.M.; Fukami, K.; Cooper, M.E. Diabetic Nephropathy: Where Hemodynamics Meets Metabolism. Exp. Clin. Endocrinol. Diabetes 2007, 115, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Lajer, M.; Tarnow, L.; Fleckner, J.; Hansen, B.V.; Edwards, D.G.; Parving, H.H.; Boel, E. Association of Aldose Reductase Gene Z+2 Polymorphism with Reduced Susceptibility to Diabetic Nephropathy in Caucasian Type 1 Diabetic Patients. Diabet. Med. 2004, 21, 867–873. [Google Scholar] [CrossRef]
- Zhao, H.L.; Tong, P.C.; Lai, F.M.; Tomlinson, B.; Chan, J.C. Association of Glomerulopathy with the 5′-End Polymorphism of the Aldose Reductase Gene and Renal Insufficiency in Type 2 Diabetic Patients. Diabetes 2004, 53, 2984–2991. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demaine, A.G. Polymorphisms of the Aldose Reductase Gene and Susceptibility to Diabetic Microvascular Complications. Curr. Med. Chem. 2003, 10, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J. Aldose Reductase Inhibitors and Diabetic Kidney Disease. Curr. Opin. Investig. Drugs 2010, 11, 402–417. [Google Scholar]
- Xu, M.; Chen, X.; Yan, L.; Cheng, H.; Chen, W. Association between (AC)n Dinucleotide Repeat Polymorphism at the 5′-End of the Aldose Reductase Gene and Diabetic Nephropathy: A Meta-Analysis. J. Mol. Endocrinol. 2008, 40, 243–251. [Google Scholar] [CrossRef]
- Bank, N.; Mower, P.; Aynedjian, H.S.; Wilkes, B.M.; Silverman, S. Sorbinil Prevents Glomerular Hyperperfusion in Diabetic Rats. Am. J. Physiol. 1989, 256, 1000–1006. [Google Scholar] [CrossRef]
- Tilton, R.G.; Chang, K.; Pugliese, G.; Eades, D.M.; Province, M.A.; Sherman, W.R.; Kilo, C.; Williamson, J.R. Prevention of Hemodynamic and Vascular Albumin Filtration Changes in Diabetic Rats by Aldose Reductase Inhibitors. Diabetes 1989, 38, 1258–1270. [Google Scholar] [CrossRef]
- Passariello, N.; Sepe, J.; Marrazzo, G.; De Cicco, A.; Peluso, A.; Pisano, M.C.; Sgambato, S.; Tesauro, P.; D’Onofrio, F. Effect of Aldose Reductase Inhibitor (Tolrestat) on Urinary Albumin Excretion Rate and Glomerular Filtration Rate in IDDM Sub-Jects with Nephropathy. Diabetes Care 1993, 16, 789–795. [Google Scholar] [CrossRef]
- Pedersen, M.M.; Christiansen, J.S.; Mogensen, C.E. Reduction of Glomerular Hyperfiltration in Normoalbuminuric IDDM Patients by 6 Mo of Aldose Reductase Inhibition. Diabetes 1991, 40, 527–531. [Google Scholar] [CrossRef]
- Tilton, R.G.; Kawamura, T.; Chang, K.C.; Ido, Y.; Bjercke, R.J.; Stephan, C.C.; Brock, T.A.; Williamson, J.R. Vascular Dysfunction Induced by Elevated Glucose Levels in Rats is Mediated by Vascular Endothelial Growth Factor. J. Clin. Investig. 1997, 99, 2192–2202. [Google Scholar] [CrossRef]
- McAuliffe, A.V.; Brooks, B.A.; Fisher, E.J.; Molyneaux, L.M.; Yue, D.K. Administration of Ascorbic Acid and an Aldose Reductase Inhibitor (Tolrestat) in Diabetes: Effect on Urinary Albumin Excretion. Nephron 1998, 80, 277–284. [Google Scholar] [CrossRef]
- Iso, K.; Tada, H.; Kuboki, K.; Inokuchi, T. Long-Term Effect of Epalrestat, an Aldose Reductase Inhibitor, on the Development of Incipient Diabetic Nephropathy in Type 2 Diabetic Patients. J. Diabetes Complicat. 2001, 15, 241–244. [Google Scholar] [CrossRef]
- Hamada, Y.; Nakamura, J.; Naruse, K.; Komori, T.; Kato, K.; Kasuya, Y.; Nagai, R.; Horiuchi, S.; Hotta, N. Epalrestat, an Aldose Reductase Ihibitor, Reduces the Levels of Nepsilon-(Carboxymethyl)lysine Protein Adducts and Their Pre-Cursors in Erythrocytes from Diabetic Patients. Diabetes Care 2000, 23, 1539–1544. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tahrani, A.A.; Askwith, T.; Stevens, M.J. Emerging Drugs for Diabetic Neuropathy. Expert Opin. Emerg. Drugs 2010, 15, 661–683. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.V.; Snksen, P.H.; Boulton, A.J.; Macleod, A.; Beg, M.; Bochenek, W.; Graepel, G.J.; Gonen, B. Withdrawal of the Aldose Reductase Inhibitor Tolrestat in Patients with Diabetic Neuropathy: Effect on Nerve Function. J. Diabetes Complicat. 1993, 7, 170–178. [Google Scholar] [CrossRef]
- Giugliano, D.; Acampora, R.; Marfella, R.; Di Maro, G.; De Rosa, N.; Misso, L.; Ceriello, A.; Quatraro, A.; D’Onofrio, F. Tolrestat in the Primary Prevention of Diabetic Neuropathy. Diabetes Care 1995, 18, 536–541. [Google Scholar] [CrossRef]
- Boulton, A.J.; Levin, S.; Comstock, J. A Multicentre Trial of the Aldose-Reductase Inhibitor, Tolrestat, in Patients with Symptomatic Diabetic Neuropathy. Diabetologia 1990, 33, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Macleod, A.F.; Boulton, A.J.; Owens, D.R.; Van Rooy, P.; Van Gerven, J.M.; Macrury, S.; Scarpello, J.H.; Segers, O.; Heller, S.R.; Van Der Veen, E.A. A Multicentre Trial of the Aldose-Reductase Inhibitor Tolrestat, in Patients with Symptomatic Diabetic Peripheral Neuropathy. Diabete Metab. 1992, 18, 14–20. [Google Scholar] [PubMed]
- Bril, V.; Hirose, T.; Tomioka, S.; Buchanan, R. Ranirestat for the Management of Diabetic Sensorimotor Polyneuropathy. Diabetes Care 2009, 32, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, K.; Narabayashi, I.; Tamura, K.; Nakatani, Y.; Saika, Y.; Onishi, S.; Kariyone, S. Effects of Aldose Reductase Inhibitor and Vitamin B12 on Myocardial Uptake of Iodine-123 Metaiodobenzylguanidine in Patients with Non-Insulin-Dependent Diabetes mellitus. Eur. J. Nucl. Med. 1998, 25, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.T.; Hazemann, I.; Mitschler, A.; Carbone, V.; Joachimiak, A.; Ginell, S.; Podjarny, A.; El-Kabbani, O. Unusual Binding Mode of the 2S4R Stereoisomer of the Potent Aldose Reductase Cyclic Imide Inhibitor Fidarestat (2S4S) in the 15 K Crystal Structure of the Ternary Complex Refined at 0.78 a Resolution: Implications for the Inhibition Mechanism. J. Med. Chem. 2008, 51, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Pacher, P.; Szabo, C.; Zsengeller, Z.; Hirooka, H.; Stevens, M.J.; Yorek, M.A. Aldose Reductase Inhibition Counteracts Oxidative-Nitrosative Stress and poly(ADP-Ribose) Polymerase Activation in Tissue Sites for Diabetes Complications. Diabetes 2005, 54, 234–242. [Google Scholar] [CrossRef]
- Hotta, N.; Akanuma, Y.; Kawamori, R.; Matsuoka, K.; Oka, Y.; Shichiri, M.; Toyota, T.; Nakashima, M.; Yoshimura, I.; Sakamoto, N.; et al. Long-Term Clinical Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy: The 3-Year, Multi-Center, Comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 2006, 29, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Hotta, N.; Kawamori, R.; Fukuda, M.; Shigeta, Y. Long-Term Clinical Effects of Epalrestat, an Aldose Reductase Inhibitor, on Progression of Diabetic Neuropathy and Other Micro-Vascular Complications: Multivariate Epidemiological Analysis Based on Patient Background Factors and Severity of Diabetic. Diabet. Med. A J. Br. Diabet. Assoc. 2012, 29, 1529–1533. [Google Scholar] [CrossRef]
- Sima, A.A.; Greene, D.A.; Brown, M.B.; Hohman, T.C.; Hicks, D.; Graepel, G.J.; Bochenek, W.J.; Beg, M.; Gonen, B. Effect of Hyperglycemia and the Aldose Reductase Inhibitor Tolrestat on Sural Nerve Biochemistry and Morphometry in Advanced Diabetic Peripheral Polyneuropathy. J. Diabetes Complicat. 1993, 7, 157–169. [Google Scholar] [CrossRef][Green Version]
- Rakowitz, D.; Piccolruaz, G.; Pirker, C.; Matuszczak, B. Novel Aldose Reductase Inhibitors Derived From 6-[[(Diphenylmethylene)amino]oxy]hexanoic Acid. Arch. Pharm. 2007, 340, 202–208. [Google Scholar] [CrossRef]
- Mizukami, H.; Osonoi, S.; Takaku, S.; Yamagishi, S.I.; Ogasawara, S.; Sango, K.; Chung, S.; Yagihashi, S. Role of Glucosamine in Development of Diabetic Neuropathy Independent of the Aldose Reductase Pathway. Brain Commun. 2020, 2, fcaa168. [Google Scholar] [CrossRef]
- Maccari, R.; Ottana, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef]
- Ramana, K.V.; Willis, M.S.; White, M.D.; Horton, J.W.; DiMaio, J.M.; Srivastava, D.; Bhatnagar, A.; Srivastava, S.K. Endotoxin-Induced Cardiomyopathy and Systemic Inflammation in Mice Is Prevented by Aldose Reductase Inhibition. Circulation 2006, 114, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.B.; Srivastava, S.K.; Ramana, K.V. Anti-Inflammatory Effect of Aldose Reductase Inhibition in Murine Polymicrobial Sepsis. Cytokine 2009, 48, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Ramana, K.V.; Singhal, S.S.; Awasthi, S.; Srivastava, S.K. Aldose Reductase Regulates Growth Factor-Induced Cyclooxygenase-2 Expression and Prostaglandin E2 Production in Human Colon Cancer Cells. Cancer Res. 2006, 66, 9705–9713. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Tammali, R.; Srivastava, S.K. Inhibition of Aldose Reductase Prevents Growth Factor-Induced G1-S Phase Transition through the AKT/Phosphoinositide 3-Kinase/E2F-1 Pathway in Human Colon Cancer Cells. Mol. Cancer Ther. 2010, 9, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Reddy, A.B.; Ramana, K.V.; Petrash, J.M.; Srivastava, S.K. Aldose Reductase Deficiency in Mice Prevents Azoxymethane-Induced Colonic Preneoplastic Aberrant Crypt Foci Formation. Carcinogenesis 2009, 30, 799–807. [Google Scholar] [CrossRef]
- Saxena, A.; Shoeb, M.; Tammali, R.; Ramana, K.V.; Srivastava, S.K. Aldose Reductase Inhibition Suppresses Azoxymethane-Induced Colonic Premalignant Lesions in C57BL/KsJ-DB/DB Mice. Cancer Lett. 2014, 355, 141–147. [Google Scholar] [CrossRef]
- Saraswat, M.; Mrudula, T.; Kumar, P.U.; Suneetha, A.; Rao Rao, T.S.; Srinivasulu, M.; Reddy, B. Overexpression of Aldose Reductase in Human Cancer Tissues. Med. Sci. Monit. 2006, 12, 525–529. [Google Scholar]
- Lee, K.W.; Ko, B.C.; Jiang, Z.; Cao, D.; Chung, S.S. Overexpression of Aldose Reductase in Liver Cancers May Contribute to Drug Resistance. Anticancer. Drugs 2001, 12, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Tammali, R.; Ramana, K.V.; Srivastava, S.K. Aldose Reductase Inhibition Prevents Colon Cancer Growth by Restoring Phosphatase and Tensin Homolog through Modulation of miR-21 and FOXO3a. Antioxid. Redox Signal. 2013, 18, 1249–1262. [Google Scholar] [CrossRef]
- Bradley, J.; Ju, M.; Robinson, G.S. Combination Therapy for the Treatment of Ocular Neovascularization. Angiogenesis 2007, 10, 141–148. [Google Scholar] [CrossRef]
- Pandey, S.; Srivastava, S.K.; Ramana, K.V. A Potential Therapeutic Role for Aldose Reductase Inhibitors in the Treatment of Endotoxin-Related Inflammatory Diseases. Expert Opin. Investig. Drugs 2012, 21, 329–339. [Google Scholar] [CrossRef]
- Alexiou, P.; Pegklidou, K.; Chatzopoulou, M.; Nicolaou, I.; Demopoulos, V.J. Aldose Reductase Enzyme and Its Implication to Major Health Problems of the 21st Century. Curr. Med. Chem. 2009, 16, 734–752. [Google Scholar] [CrossRef]
- Demir, Y.; Taslimi, P.; Koçyiğit, Ü.M.; Akkuş, M.; Özaslan, M.S.; Duran, H.E.; Budak, Y.; Tüzün, B.; Gürdere, M.B.; Ceylan, M.; et al. Determination of the Inhibition Profiles of Pyrazolyl-Thiazole Derivatives Against Aldose Reductase and α-Glycosidase and Molecular Docking Studies. Arch. Der Pharm. 2020, 353, e2000118. [Google Scholar] [CrossRef]
- Zaher, N.; Nicolaou, I.; Demopoulos, V.J. Pyrrolylbenzothiazole Derivatives as Aldose Reductase Inhibitors. J. Enzym. Inhib. Med. Chem. 2002, 17, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Backonja, M.; Beydoun, A.; Edwards, K.R.; Schwartz, S.L.; Fonseca, V.; Hes, M.; LaMoreaux, L.; Garofalo, E. Gabapentin for the Symptomatic Treatment of Painful Neuropathy in Patients with Diabetes mellitus: A Randomized Controlled Trial. Jama 1998, 280, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T. The Role of Polyols in the Pathophysiology of Hypergalactosemia. Eur. J. Pediatrics 1995, 154, 53–64. [Google Scholar] [CrossRef]
- Andres-Hernando, A.; Li, N.; Cicerchi, C.; Inaba, S.; Chen, W.; Roncal-Jimenez, C.; Le, M.T.; Wempe, M.F.; Milagres, T.; Ishimoto, T.; et al. Protective Role of Fructokinase Blockade in the Pathogenesis of Acute Kidney Injury in Mice. Nat. Commun. 2017, 8, 14181. [Google Scholar] [CrossRef]
- García-Arroyo, F.E.; Tapia, E.; Blas-Marron, M.G.; Gonzaga, G.; Silverio, O.; Cristóbal, M.; Osorio, H.; Arellano-Buendía, A.S.; Zazueta, C.; Aparicio-Trejo, O.E.; et al. Vasopressin Mediates the Renal Damage Induced by Limited Fructose Rehydration in Recurrently Dehydrated Rats. Int. J. Biol. Sci. 2017, 13, 961–975. [Google Scholar] [CrossRef]
- Hasuike, Y.; Nakanishi, T.; Otaki, Y.; Nanami, M.; Tanimoto, T.; Taniguchi, N.; Takamitsu, Y. Plasma 3-Deoxyglucosone Elevation in Chronic Renal Failure Is Associated with Increased Aldose Reductase in Erythrocytes. Am. J. Kidney Dis. 2002, 40, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Tiwari, A.K.; Kumar, K.M.; Ammini, A.C.; Gupta, A.; Gupta, R.; Thelma, B.K. Association Analysis of ADPRT1, AKR1B1, RAGE, GFPT2 and PAI-1 Gene Polymorphisms with Chronic Renal Insufficiency Among Asian Indians with Type-2 Diabetes. BMC Med. Genet. 2010, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mizukami, H.; Kamata, K.; Inaba, W.; Kato, N.; Hibi, C.; Yagihashi, S. Amelioration of Acute Kidney Injury in Lipopolysaccharide-Induced Systemic Inflammatory Response Syndrome by an Aldose Reductase Inhibitor, Fidarestat. PLoS ONE 2012, 7, e30134. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Mizukami, H.; Ogasawara, S.; Yamagishi, S.; Nukada, H.; Kato, N.; Hibi, C.; Chung, S.; Chung, S. The Role of the Polyol Pathway in Acute Kidney Injury Caused by Hindlimb Ischaemia in Mice. J. Pathol. 2010, 220, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Zhang, R.; Anand, P.; Stomberski, C.T.; Qian, Z.; Hausladen, A.; Wang, L.; Rhee, E.P.; Parikh, S.M.; Karumanchi, S.A.; et al. Metabolic Reprogramming by the S-Nitroso-CoA Reductase System Protects Against Kidney Injury. Nature 2019, 565, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Tammali, R.; Reddy, A.B.; Saxena, A.; Rychahou, P.G.; Evers, B.M.; Qiu, S.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Inhibition of Aldose Reductase Prevents Colon Cancer Metastasis. Carcinogenesis 2011, 32, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.X.; Yuan, Y.W.; Cai, C.F.; Shen, D.Y.; Chen, M.L.; Ye, F.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; Zhang, W.; et al. Aldose Reductase Interacts with AKT1 to Augment Hepatic AKT/mTOR Signaling and Promote Hepatocarcinogenesis. Oncotarget 2017, 8, 66987–67000. [Google Scholar] [CrossRef]
- Jamialahmadi, K.; Azghandi, M.; Javadmanesh, A.; Zardadi, M.; Shams Davodly, E.; Kerachian, M.A. A DNA Methylation Panel for High Performance Detection of Colorectal Cancer. Cancer Genet. 2021, 252, 64–72. [Google Scholar] [CrossRef]
- Huang, L.; He, R.; Luo, W.; Zhu, Y.S.; Li, J.; Tan, T.; Zhang, X.; Hu, Z.; Luo, D. Aldo-Keto Reductase Family 1 Member B10 Inhibitors: Potential Drugs for Cancer Treatment. Recent Pat. Anti-Cancer Drug Discov. 2016, 11, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Krishack, P.A.; Cao, D. Role of Aldo-Keto Reductases in Development of Prostate and Breast Cancer. Front. Biosci. 2006, 11, 2767–2773. [Google Scholar] [CrossRef][Green Version]
- Qu, J.; Liu, X.; Li, J.; Gong, K.; Duan, L.; Luo, W.; Luo, D. AKR1B10 Promotes Proliferation of Breast Cancer Cells by Activating Wnt/β-Catenin Pathway. Chin. J. Cell. Mol. Immunol. 2019, 35, 1094–1100. [Google Scholar]
- Sonowal, H.; Pal, P.; Shukla, K.; Saxena, A.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Inhibitor, Fidarestat Prevents Doxorubicin-Induced Endothelial Cell Death and Dysfunction. Biochem. Pharmacol. 2018, 150, 181–190. [Google Scholar] [CrossRef]
- Van Weverwijk, A.; Koundouros, N.; Iravani, M.; Ashenden, M.; Gao, Q.; Poulogiannis, G.; Jungwirth, U.; Isacke, C.M. Metabolic Adaptability in Metastatic Breast Cancer by AKR1B10-Dependent Balancing of Glycolysis and Fatty Acid Oxidation. Nat. Commun. 2019, 10, 2698. [Google Scholar] [CrossRef]
- Chatzopoulou, M.; Alexiou, P.; Kotsampasakou, E.; Demopoulos, V.J. Novel Aldose Reductase Inhibitors: A Patent Survey (2006–Present). Expert Opin. Ther. Pat. 2012, 22, 1303–1323. [Google Scholar] [CrossRef]
- Ramunno, A.; Cosconati, S.; Sartini, S.; Maglio, V.; Angiuoli, S.; La Pietra, V.; Di Maro, S.; Giustiniano, M.; La Motta, C.; Da Settimo, F.; et al. Progresses in the Pursuit of Aldose Reductase Inhibitors: The Structure-Based Lead Optimization Step. Eur. J. Med. Chem. 2012, 51, 216–226. [Google Scholar] [CrossRef]
- Cumbie, B.C.; Hermayer, K.L. Current Concepts in Targeted Therapies for the Pathophysiology of Diabetic Microvascular Complications. Vasc. Health Risk Manag. 2007, 3, 823–832. [Google Scholar] [PubMed]




| Categories (Types/Subtypes) | AR Inhibitors | Reference(s) |
|---|---|---|
| Naturally Occurring |
| [21,22,23] |
| Flavanone Glucoside |
| [24,25,26,27,28] |
| Alkaloids |
| [29,30,31,32,33,34,35,36,37,38,39,40,41,42,43] |
| Spirohydantoin |
| [44,45,46,47,48,49,50,51,52] |
| Acetic Acid |
| [53,54,55,56,57,58,59,60,61] |
| Carboxyl Acid |
| [62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90] |
| Flavanoids |
| [29,37,91,92,93,94,95,96,97,98] |
| Spiroscinimide |
| [50,61,81,82,83,87,88,90,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. https://doi.org/10.3390/metabo11100655
Singh M, Kapoor A, Bhatnagar A. Physiological and Pathological Roles of Aldose Reductase. Metabolites. 2021; 11(10):655. https://doi.org/10.3390/metabo11100655
Chicago/Turabian StyleSingh, Mahavir, Aniruddh Kapoor, and Aruni Bhatnagar. 2021. "Physiological and Pathological Roles of Aldose Reductase" Metabolites 11, no. 10: 655. https://doi.org/10.3390/metabo11100655
APA StyleSingh, M., Kapoor, A., & Bhatnagar, A. (2021). Physiological and Pathological Roles of Aldose Reductase. Metabolites, 11(10), 655. https://doi.org/10.3390/metabo11100655