
Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome


Abstract

1. Introduction
2. Results
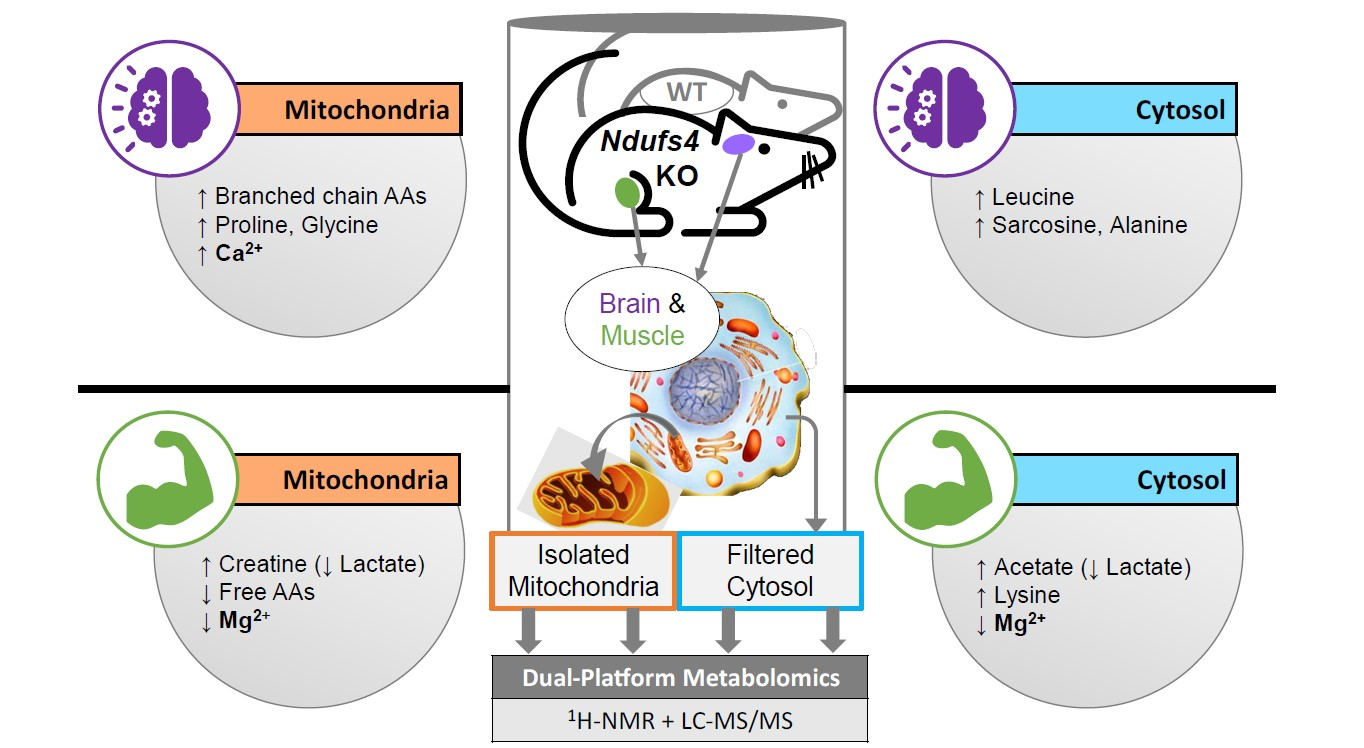
2.1. Experimental Rationale and Data Quality
2.2. Metabolome Coverage and Data Clean-Up
2.3. Compartment-Specific, Significant Ndufs4 KO Metabolite Alterations
2.4. Amino Acids Accumulate in Whole-Brain CI-Deficient Mitochondria to a Greater Extent than Cytosolic or Region-Specific Metabolomics Can Fully Elucidate
2.5. Differential Depletion of Quadriceps Mitochondrial and Cytosolic Metabolites Related to CID Muscle Metabolic Reprogramming
2.6. First Report of Tissue- and Compartment-Specific Bivalent Cation Measuremnt in Ndufs4 Mice Highlights Role of Metal Homeostasis in Integrated Stress Responses
3. Discussion
3.1. Contribution towards In Vivo Ndufs4 Model and In Vitro CID Knowledge Base
3.2. Limitations and Future Directions
4. Materials and Methods
4.1. Animals and Sampling
4.2. Mitochondrial Isolation and Cytosol Preparation
4.3. Metabolomics Group Structure and Preparation for Multiplatform Studies
4.3.1. Internal Standards and Quality Control Samples
4.3.2. Untargeted 1H-NMR Spectroscopy
4.3.3. LC-MS/MS Amino Acid and Acylcarnitine Profiling
4.3.4. Data Processing
4.3.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Houten, B.V.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, Energetics, Epigenetics, and Cellular Responses to Stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef]
- Aouacheria, A.; Baghdiguian, S.; Lamb, H.M.; Huska, J.D.; Pineda, F.J.; Hardwick, J.M. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 2017, 109, 141–161. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Wasilewski, M.; Chojnacka, K.; Chacinska, A. Protein trafficking at the crossroads to mitochondria. Biochim. Biophys. Acta Mol. Cell Res 2017, 1864, 125–137. [Google Scholar] [CrossRef]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular Dysfunctions of Mitochondria-Associated Membranes (MAMs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Suomalainen, A. Mechanisms of mitochondrial diseases. Ann. Med. 2012, 44, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Triepels, R.; Van Den Heuvel, L.; Trijbels, J.; Smeitink, J. Respiratory chain complex I deficiency. Am. J. Med. Genet. 2001, 106, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [PubMed]
- Lake, N.J.; Bird, M.J.; Isohanni, P.; Paetau, A. Leigh syndrome: Neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 2015, 74, 482–492. [Google Scholar] [CrossRef]
- Quintana, A.; Kruse, S.E.; Kapur, R.P.; Sanz, E.; Palmiter, R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10996–11001. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Priori, R.; Scrivo, R.; Brandt, J.; Valerio, M.; Casadei, L.; Valesini, G.; Manetti, C. Metabolomics in rheumatic diseases: The potential of an emerging methodology for improved patient diagnosis, prognosis, and treatment efficacy. Autoimmun. Rev. 2013, 12, 1022–1030. [Google Scholar] [CrossRef]
- Nagrath, D.; Caneba, C.; Karedath, T.; Bellance, N. Metabolomics for mitochondrial and cancer studies. Biochim. Biophys. Acta 2011, 1807, 650–663. [Google Scholar] [CrossRef]
- Kruse, S.E.; Watt, W.C.; Marcinek, D.J.; Kapur, R.P.; Schenkman, K.A.; Palmiter, R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008, 7, 312–320. [Google Scholar] [CrossRef]
- Leong, D.W.; Komen, J.C.; Hewitt, C.A.; Arnaud, E.; McKenzie, M.; Phipson, B.; Bahlo, M.; Laskowski, A.; Kinkel, S.A.; Davey, G.M.; et al. Proteomic and metabolomic analyses of mitochondrial complex I-deficient mouse model generated by spontaneous B2 short interspersed nuclear element (SINE) insertion into NADH dehydrogenase (ubiquinone) Fe-S protein 4 (Ndufs4) gene. J. Biol. Chem. 2012, 287, 20652–20663. [Google Scholar] [CrossRef]
- Esterhuizen, K.; van der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef]
- Johnson, S.C.; Kayser, E.B.; Bornstein, R.; Stokes, J.; Bitto, A.; Park, K.Y.; Pan, A.; Sun, G.; Raftery, D.; Kaeberlein, M.; et al. Regional metabolic signatures in the Ndufs4(KO) mouse brain implicate defective glutamate/alpha-ketoglutarate metabolism in mitochondrial disease. Mol. Genet. Metab. 2020, 130, 118–132. [Google Scholar] [CrossRef]
- Terburgh, K.; Coetzer, J.; Lindeque, J.Z.; van der Westhuizen, F.H.; Louw, R. Aberrant BCAA and glutamate metabolism linked to regional neurodegeneration in a mouse model of Leigh syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166082. [Google Scholar] [CrossRef]
- Emmerzaal, T.L.; Preston, G.; Geenen, B.; Verweij, V.; Wiesmann, M.; Vasileiou, E.; Gruter, F.; de Groot, C.; Schoorl, J.; de Veer, R.; et al. Impaired mitochondrial complex I function as a candidate driver in the biological stress response and a concomitant stress-induced brain metabolic reprogramming in male mice. Transl. Psychiatry 2020, 10, 176. [Google Scholar] [CrossRef]
- Terburgh, K.; Lindeque, Z.; Mason, S.; van der Westhuizen, F.; Louw, R. Metabolomics of Ndufs4−/− skeletal muscle: Adaptive mechanisms converge at the ubiquinone-cycle. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 98–106. [Google Scholar] [CrossRef]
- Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 2008, 39, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivela, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e6. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics—Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Johnson, M.A.; Vidoni, S.; Durigon, R.; Pearce, S.F.; Rorbach, J.; He, J.; Brea-Calvo, G.; Minczuk, M.; Reyes, A.; Holt, I.J.; et al. Amino acid starvation has opposite effects on mitochondrial and cytosolic protein synthesis. PLoS ONE 2014, 9, e93597. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkila, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjosund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, G. Recent advances in using mass spectrometry for mitochondrial metabolomics and lipidomics—A review. Anal. Chim. Acta 2018, 1037, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef]
- Wiechert, W. 13C metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Gravel, S.P.; Andrzejewski, S.; Avizonis, D.; St-Pierre, J. Stable isotope tracer analysis in isolated mitochondria from mammalian systems. Metabolites 2014, 4, 166–183. [Google Scholar] [CrossRef]
- Roede, J.R.; Park, Y.; Li, S.; Strobel, F.H.; Jones, D.P. Detailed mitochondrial phenotyping by high resolution metabolomics. PLoS ONE 2012, 7, e33020. [Google Scholar] [CrossRef]
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J. Biol. Chem. 2018, 293, 7508–7516. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Roede, J.R.; Orr, M.; Liang, Y.; Jones, D.P. Integrated redox proteomics and metabolomics of mitochondria to identify mechanisms of cd toxicity. Toxicol. Sci. 2014, 139, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Han, X.; Cheng, H.; Seyfried, T.N. In vitro growth environment produces lipidomic and electron transport chain abnormalities in mitochondria from non-tumorigenic astrocytes and brain tumours. ASN Neuro 2009, 1, AN20090011. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Freinkman, E.; Wang, T.; Birsoy, K.; Sabatini, D.M. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 2016, 166, 1324–1337.e11. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Lindau, C.; Lagies, S.; Wiedemann, N.; Kammerer, B. Metabolic profiling of isolated mitochondria and cytoplasm reveals compartment-specific metabolic responses. Metabolomics 2018, 14, 59. [Google Scholar] [CrossRef]
- Bayraktar, E.C.; Baudrier, L.; Ozerdem, C.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Cangelosi, A.L.; Sabatini, D.M.; Birsoy, K.; et al. MITO-Tag Mice enable rapid isolation and multimodal profiling of mitochondria from specific cell types in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 303–312. [Google Scholar] [CrossRef] [PubMed]
- van der Walt, G.; Louw, R. Novel mitochondrial and cytosolic purification pipeline for compartment-specific metabolomics in mammalian disease model tissues. Metabolomics 2020, 16, 78. [Google Scholar] [CrossRef]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G* Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Mason, S.; Terburgh, K.; Louw, R. Miniaturized 1H-NMR method for analyzing limited-quantity samples applied to a mouse model of Leigh disease. Metabolomics 2018, 14, 74. [Google Scholar] [CrossRef]
- Mels, C.; Jansen van Rensburg, P.; van der Westhuizen, F.H.; Pretorius, P.J.; Erasmus, E. Increased excretion of C4-carnitine species after a therapeutic acetylsalicylic acid dose: Evidence for an inhibitory effect on short-chain fatty acid metabolism. Int. Sch. Res. Not. 2011, 2011, 851870. [Google Scholar] [CrossRef]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- US FDA. Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV): Rockville, MD, USA, 2001. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 1 September 2021).
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Akobeng, A.K. Understanding type I and type II errors, statistical power and sample size. Acta Paediatr. 2016, 105, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Bifari, F.; Nisoli, E. Branched-chain amino acids differently modulate catabolic and anabolic states in mammals: A pharmacological point of view. Br. J. Pharmacol. 2017, 174, 1366–1377. [Google Scholar] [CrossRef]
- Arany, Z.; Neinast, M. Branched Chain Amino Acids in Metabolic Disease. Curr. Diabetes Rep. 2018, 18, 76. [Google Scholar] [CrossRef]
- Oka, K.; Ohya-Shimada, W.; Mizuno, S.; Nakamura, T. Up-regulation of cyclin-E1 via proline-mTOR pathway is responsible for HGF-mediated G1/S progression in the primary culture of rat hepatocytes. Biochem. Biophys. Res. Commun. 2013, 435, 120–125. [Google Scholar] [CrossRef]
- Krishnan, N.; Dickman, M.B.; Becker, D.F. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med. 2008, 44, 671–681. [Google Scholar] [CrossRef]
- McDonald, A.E.; Pichaud, N.; Darveau, C.A. “Alternative” fuels contributing to mitochondrial electron transport: Importance of non-classical pathways in the diversity of animal metabolism. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2018, 224, 185–194. [Google Scholar] [CrossRef]
- Razak, M.A.; Begum, P.S.; Viswanath, B.; Rajagopal, S. Multifarious Beneficial Effect of Nonessential Amino Acid, Glycine: A Review. Oxidative Med. Cell. Longev. 2017, 2017, 1716701. [Google Scholar] [CrossRef]
- Wang, W.; Wu, Z.; Dai, Z.; Yang, Y.; Wang, J.; Wu, G. Glycine metabolism in animals and humans: Implications for nutrition and health. Amino Acids 2013, 45, 463–477. [Google Scholar] [CrossRef]
- Yang, L.; Garcia Canaveras, J.C.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821.e6. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirk, K.; Shurubor, Y.I.; Zhao, D.; Arreguin, A.J.; Shahi, I.; Valsecchi, F.; Primiano, G.; Calder, E.L.; Carelli, V. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018, 27, 1007–1025.e5. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Park, S.J.; Stamatakou, E.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat. Commun. 2020, 11, 3148. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020, 35, 101454. [Google Scholar] [CrossRef]
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008, 582, 3569–3576. [Google Scholar] [CrossRef]
- Young, A.; Oldford, C.; Mailloux, R.J. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol. 2020, 28, 101339. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Schwörer, S.; Berisa, M.; Violante, S.; Qin, W.; Zhu, J.; Hendrickson, R.C.; Cross, J.R.; Thompson, C.B. Proline biosynthesis is a vent for TGFβ-induced mitochondrial redox stress. EMBO J. 2020, 39, e103334. [Google Scholar] [CrossRef]
- Sarabhai, T.; Roden, M. Hungry for your alanine: When liver depends on muscle proteolysis. J. Clin. Investig. 2019, 129, 4563–4566. [Google Scholar] [CrossRef]
- Holeček, M. Histidine in Health and Disease: Metabolism, Physiological Importance, and Use as a Supplement. Nutrients 2020, 12, 848. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Shao, W.; Huang, C.; Wu, C.-J.; Jiang, B.; Lin, D. Metabolic derangements of skeletal muscle from a murine model of glioma cachexia. Skelet. Muscle 2019, 9, 3. [Google Scholar] [CrossRef]
- Ross-Inta, C.; Tsai, C.Y.; Giulivi, C. The mitochondrial pool of free amino acids reflects the composition of mitochondrial DNA-encoded proteins: Indication of a post- translational quality control for protein synthesis. Biosci. Rep. 2008, 28, 239–249. [Google Scholar] [CrossRef]
- Mathuthu, E.; Janse van Rensburg, A.; Du Plessis, D.; Mason, S. EDTA as a chelating agent in quantitative 1H-NMR of biologically important ions. Biochem. Cell Biol. 2021, 99, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hafer, E.; Holzgrabe, U.; Kraus, K.; Adams, K.; Hook, J.M.; Diehl, B. Qualitative and quantitative 1H NMR spectroscopy for determination of divalent metal cation concentration in model salt solutions, food supplements, and pharmaceutical products by using EDTA as chelating agent. Magn. Reson. Chem. 2020, 58, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Hornig-Do, H.T.; Gunther, G.; Bust, M.; Lehnartz, P.; Bosio, A.; Wiesner, R.J. Isolation of functional pure mitochondria by superparamagnetic microbeads. Anal. Biochem. 2009, 389, 1–5. [Google Scholar] [CrossRef]
- Koshenov, Z.; Oflaz, F.E.; Hirtl, M.; Pilic, J.; Bachkoenig, O.A.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Rost, R.; Malli, R.; Graier, W.F. Sigma-1 Receptor Promotes Mitochondrial Bioenergetics by Orchestrating ER Ca2+ Leak during Early ER Stress. Metabolites 2021, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Veronese, N.; Dominguez, L.J. Magnesium in Aging, Health and Diseases. Nutrients 2021, 13, 463. [Google Scholar] [CrossRef]
- Huskisson, E.; Maggini, S.; Ruf, M. The role of vitamins and minerals in energy metabolism and well-being. J. Int. Med. Res. 2007, 35, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Heidari, R.; Ahmadi, A.; Mohammadi, H.; Ommati, M.M.; Azarpira, N.; Niknahad, H. Mitochondrial dysfunction and oxidative stress are involved in the mechanism of methotrexate-induced renal injury and electrolytes imbalance. Biomed. Pharmacother. 2018, 107, 834–840. [Google Scholar] [CrossRef]
- Alam, M.T.; Manjeri, G.R.; Rodenburg, R.J.; Smeitink, J.A.; Notebaart, R.A.; Huynen, M.; Willems, P.H.; Koopman, W.J. Skeletal muscle mitochondria of NDUFS4−/− mice display normal maximal pyruvate oxidation and ATP production. Biochim. Biophys. Acta 2015, 1847, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, D.A.; Kreider, R.B.; Stout, J.R.; Forero, D.A.; Kerksick, C.M.; Roberts, M.D.; Rawson, E.S. Metabolic Basis of Creatine in Health and Disease: A Bioinformatics-Assisted Review. Nutrients 2021, 13, 1238. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Szibor, M.; Gizatullina, Z.; Gainutdinov, T.; Endres, T.; Debska-Vielhaber, G.; Kunz, M.; Karavasili, N.; Hallmann, K.; Schreiber, F.; Bamberger, A.; et al. Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 2020, 295, 4383–4397. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M. Interactions in the Metabolism of Glutamate and the Branched-Chain Amino Acids and Ketoacids in the CNS. Neurochem. Res. 2017, 42, 10–18. [Google Scholar] [CrossRef]
- Neurauter, G.; Schrocksnadel, K.; Scholl-Burgi, S.; Sperner-Unterweger, B.; Schubert, C.; Ledochowski, M.; Fuchs, D. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr. Drug Metab. 2008, 9, 622–627. [Google Scholar] [CrossRef]
- Nikkanen, J.; Forsstrom, S.; Euro, L.; Paetau, I.; Kohnz, R.A.; Wang, L.; Chilov, D.; Viinamaki, J.; Roivainen, A.; Marjamaki, P.; et al. Mitochondrial DNA Replication Defects Disturb Cellular dNTP Pools and Remodel One-Carbon Metabolism. Cell Metab. 2016, 23, 635–648. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Harwood, C.L.; Prajapati, P.; Springer, J.E.; Saatman, K.E.; Sullivan, P.G. Fractionated mitochondrial magnetic separation for isolation of synaptic mitochondria from brain tissue. Sci. Rep. 2019, 9, 9656. [Google Scholar] [CrossRef]
- McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Nelson, M.A.M.; Kew, K.A.; Wooten, A.R.; Fisher-Wellman, K.H. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 2020, 10, 17599. [Google Scholar] [CrossRef] [PubMed]
- Ruoppolo, M.; Caterino, M.; Albano, L.; Pecce, R.; Di Girolamo, M.G.; Crisci, D.; Costanzo, M.; Milella, L.; Franconi, F.; Campesi, I. Targeted metabolomic profiling in rat tissues reveals sex differences. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Barrington, W.; Threadgill, D.; Dearth, S.; Campagna, S.; Saxton, A.; Voy, B. Gene, Sex and Diet Interact to Control the Tissue Metabolome. FASEB J. 2016, 30, 127.2. [Google Scholar] [CrossRef]
- Miller, H.C.; Louw, R.; Mereis, M.; Venter, G.; Boshoff, J.-D.; Mienie, L.; Van Reenen, M.; Venter, M.; Lindeque, J.Z.; Domínguez-Martínez, A. Metallothionein 1 overexpression does not protect against mitochondrial disease pathology in Ndufs4 knockout mice. Mol. Neurobiol. 2021, 58, 243–262. [Google Scholar] [CrossRef]
- Dona, A.C.; Kyriakides, M.; Scott, F.; Shephard, E.A.; Varshavi, D.; Veselkov, K.; Everett, J.R. A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Comput. Struct. Biotechnol. J. 2016, 14, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quadriceps Muscle | |||||||
|---|---|---|---|---|---|---|---|
| Cytosol | Mitochondria | ||||||
| Metabolite (Level 1) | KO ∆ (vs. WT) | p | D | KO ∆ (vs. WT) | p | D | Platform |
| Lactic acid | ↓ | 0.04 | 1.2 | ↓ | 0.04 | 1.1 | 1H-NMR |
| Mg(II)-EDTA | ↓ | 0.10 | 0.9 | ↓ | 0.09 | 0.9 | 1H-NMR |
| Threonine | N.Q. | ↓ | 0.04 | 1.1 | LC-MS/MS | ||
| Glycine | N.S. | ↓ | 0.05 | 1.1 | LC-MS/MS | ||
| Creatine | N.S. | ↑ | 0.05 | 1.0 | 1H-NMR | ||
| Histidine | N.S. | ↓ | 0.07 | 1.0 | LC-MS/MS | ||
| Tyrosine | N.S. | ↓ | 0.07 | 1.0 | LC-MS/MS | ||
| Ornithine | N.D. | ↓ | 0.08 | 1.0 | LC-MS/MS | ||
| Leucine | N.S. | ↓ | 0.10 | 0.9 | LC-MS/MS | ||
| Hydroxyproline | ↓ | 0.01 | 1.8 | N.Q. | LC-MS/MS | ||
| Citrulline | ↓ | 0.07 | 1.0 | N.S. | LC-MS/MS | ||
| Acetylcarnitine | ↑ | 0.09 | 0.9 | N.S. | LC-MS/MS | ||
| Lysine | ↑ | 0.10 | 0.9 | N.S. | LC-MS/MS | ||
| Whole Brain | |||||||
| Cytosol | Mitochondria | ||||||
| Metabolite (Level 1) | KO ∆ (vs. WT) | p | D | KO ∆ (vs. WT) | p | D | Platform |
| Leucine | ↑ | 0.09 | 0.9 | ↑ | 0.03 | 1.2 | LC-MS/MS |
| Proline | N.S. | ↑ | 0.03 | 1.1 | LC-MS/MS | ||
| Phenylalanine | N.S. | ↑ | 0.03 | 1.1 | LC-MS/MS | ||
| Valine | N.S. | ↑ | 0.06 | 1.0 | LC-MS/MS | ||
| Octanoylcarnitine | N.S. | ↑ | 0.08 | 1.0 | LC-MS/MS | ||
| Glycine | N.S. | ↑ | 0.08 | 0.9 | LC-MS/MS | ||
| Ca(II)-EDTA | N.S. | ↑ | 0.09 | 0.9 | 1H-NMR | ||
| Alanine 1 | ↑ | 0.10 | 0.9 | ↑ | 0.13 | 0.8 | LC-MS/MS |
| Glutamine 1 | N.S. | ↓ | 0.12 | 0.8 | LC-MS/MS | ||
| Sarcosine 1 | ↑ | 0.11 | 0.8 | N.D. | LC-MS/MS | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Walt, G.; Lindeque, J.Z.; Mason, S.; Louw, R. Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome. Metabolites 2021, 11, 658. https://doi.org/10.3390/metabo11100658
van der Walt G, Lindeque JZ, Mason S, Louw R. Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome. Metabolites. 2021; 11(10):658. https://doi.org/10.3390/metabo11100658
Chicago/Turabian Stylevan der Walt, Gunter, Jeremie Z. Lindeque, Shayne Mason, and Roan Louw. 2021. "Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome" Metabolites 11, no. 10: 658. https://doi.org/10.3390/metabo11100658
APA Stylevan der Walt, G., Lindeque, J. Z., Mason, S., & Louw, R. (2021). Sub-Cellular Metabolomics Contributes Mitochondria-Specific Metabolic Insights to a Mouse Model of Leigh Syndrome. Metabolites, 11(10), 658. https://doi.org/10.3390/metabo11100658