
Knockout of the Amino Acid Transporter SLC6A19 and Autoimmune Diabetes Incidence in Female Non-Obese Diabetic (NOD) Mice

 , , , ,
, , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
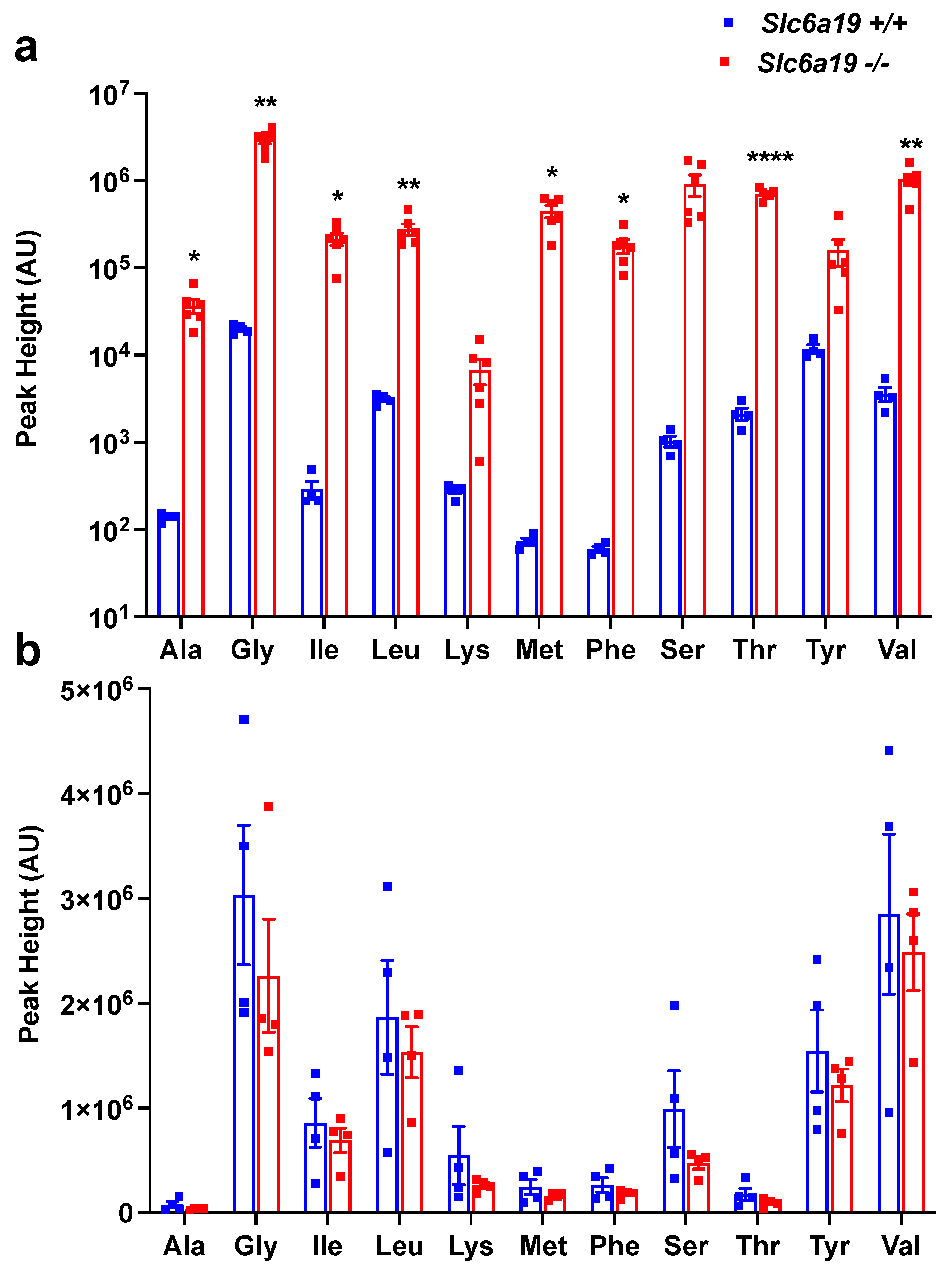
2.1. Female Slc6a19 Deficient NOD Mice Are Markedly Aminoaciduric
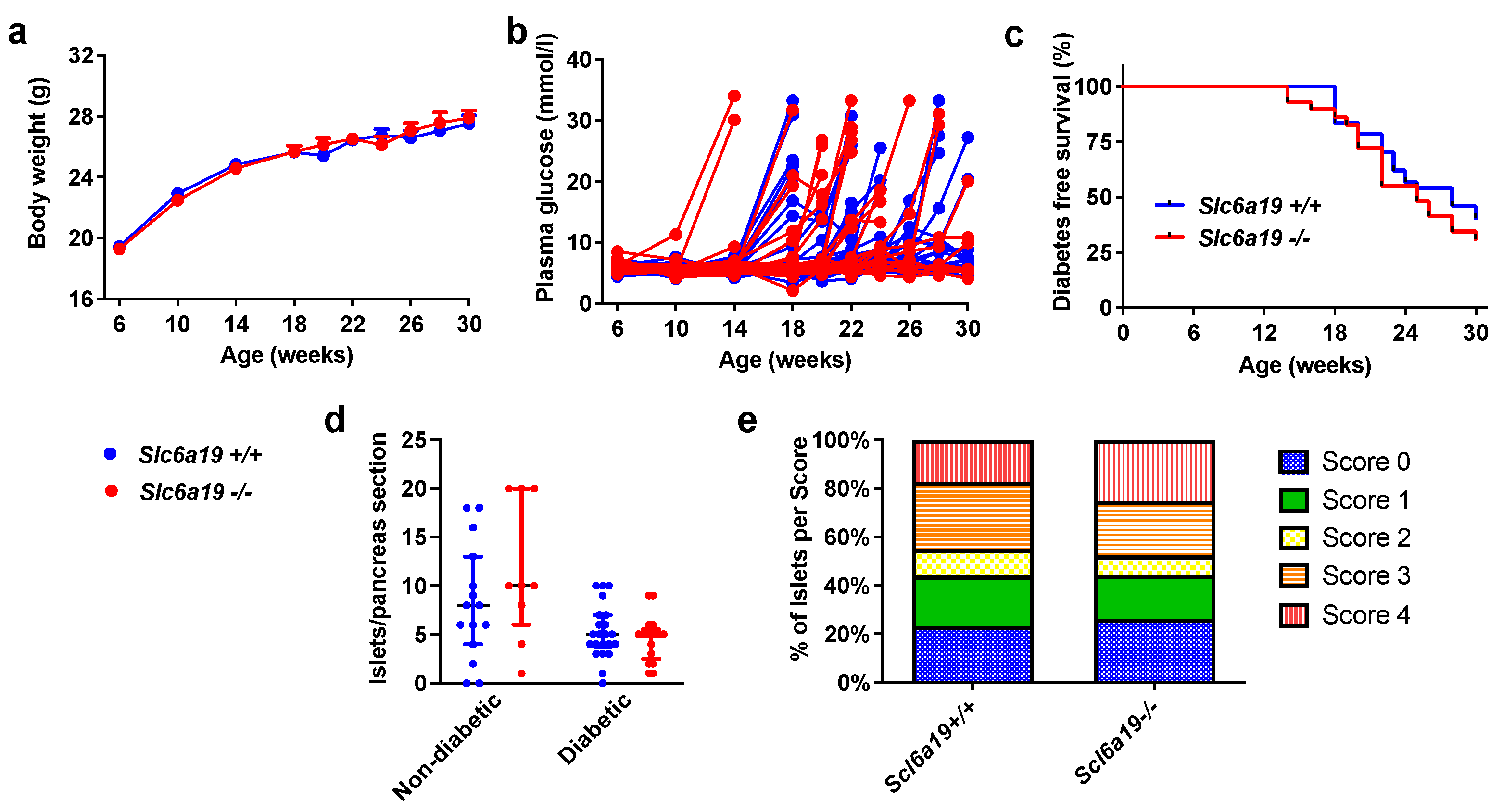
2.2. Diabetes Incidence Is Unchanged in Slc6a19 Deficient NOD Mice
2.3. Insulitis Severity Was Unaltered by Slc6a19 Deficiency in NOD Mice
3. Discussion
4. Materials and Methods
4.1. Mouse Model
4.2. Generation of CRISPR/Cas9 Edited Slc6a19 Deficient Mouse Strain
4.3. Body Weight, Glucose Measurement and Amino Acid Analyses
4.4. Pancreas Histology
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilkin, T.J. The accelerator hypothesis: Weight gain as the missing link between Type I and Type II diabetes. Diabetologia 2001, 44, 914–922. [Google Scholar] [CrossRef] [Green Version]
- Fourlanos, S.; Harrison, L.C.; Colman, P.G. The accelerator hypothesis and increasing incidence of type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 321–325. [Google Scholar] [CrossRef] [PubMed]
- von Scholten, B.J.; Kreiner, F.F.; Gough, S.C.L.; von Herrath, M. Current and future therapies for type 1 diabetes. Diabetologia 2021, 64, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Autoimmunity and oxidatively modified autoantigens. Autoimmun. Rev. 2008, 7, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Szymczak, F.; Alvelos, M.I.; Martin, F. From Pancreatic beta-Cell Gene Networks to Novel Therapies for Type 1 Diabetes. Diabetes 2021. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Divers, J.; Isom, S.; Saydah, S.; Imperatore, G.; Pihoker, C.; Marcovina, S.M.; Mayer-Davis, E.J.; Hamman, R.F.; Dolan, L.; et al. Trends in Prevalence of Type 1 and Type 2 Diabetes in Children and Adolescents in the US, 2001–2017. JAMA 2021, 326, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Ferrara-Cook, C.; Geyer, S.M.; Evans-Molina, C.; Libman, I.M.; Becker, D.J.; Gitelman, S.E.; Redondo, M.J.; Type 1 Diabetes TrialNet Study Group. Excess BMI Accelerates Islet Autoimmunity in Older Children and Adolescents. Diabetes Care 2020, 43, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Nolan, C.J.; Prentki, M. The islet beta-cell: Fuel responsive and vulnerable. Trends Endocrinol. Metab. 2008, 19, 285–291. [Google Scholar] [CrossRef]
- Wilkin, T.; Greene, S.; McCrimmon, R. Testing the accelerator hypothesis: A new approach to type 1 diabetes prevention (adAPT 1). Diabetes Obes. Metab. 2016, 18, 3–5. [Google Scholar] [CrossRef]
- Krokowski, D.; Han, J.; Saikia, M.; Majumder, M.; Yuan, C.L.; Guan, B.J.; Bevilacqua, E.; Bussolati, O.; Broer, S.; Arvan, P.; et al. A self-defeating anabolic program leads to beta-cell apoptosis in endoplasmic reticulum stress-induced diabetes via regulation of amino acid flux. J. Biol. Chem. 2013, 288, 17202–17213. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Parikh, H.; Butterworth, M.D.; Lernmark, A.; Hagopian, W.; Rewers, M.; She, J.X.; Toppari, J.; Ziegler, A.G.; Akolkar, B.; et al. Longitudinal Metabolome-Wide Signals Prior to the Appearance of a First Islet Autoantibody in Children Participating in the TEDDY Study. Diabetes 2020, 69, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Pflueger, M.; Seppanen-Laakso, T.; Suortti, T.; Hyotylainen, T.; Achenbach, P.; Bonifacio, E.; Oresic, M.; Ziegler, A.G. Age- and islet autoimmunity-associated differences in amino acid and lipid metabolites in children at risk for type 1 diabetes. Diabetes 2011, 60, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oresic, M.; Simell, S.; Sysi-Aho, M.; Nanto-Salonen, K.; Seppanen-Laakso, T.; Parikka, V.; Katajamaa, M.; Hekkala, A.; Mattila, I.; Keskinen, P.; et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J. Exp. Med. 2008, 205, 2975–2984. [Google Scholar] [CrossRef] [Green Version]
- Delovitch, T.L.; Singh, B. The nonobese diabetic mouse as a model of autoimmune diabetes: Immune dysregulation gets the NOD. Immunity 1997, 7, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Schneider, K.; Laube, H.; Linn, T. A diet enriched in protein accelerates diabetes manifestation in NOD mice. Acta Diabetol. 1996, 33, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Grapov, D.; Fahrmann, J.; Hwang, J.; Poudel, A.; Jo, J.; Periwal, V.; Fiehn, O.; Hara, M. Diabetes Associated Metabolomic Perturbations in NOD Mice. Metabolomics 2015, 11, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Seow, H.F.; Broer, S.; Broer, A.; Bailey, C.G.; Potter, S.J.; Cavanaugh, J.A.; Rasko, J.E. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat. Genet. 2004, 36, 1003–1007. [Google Scholar] [CrossRef]
- Jiang, Y.; Rose, A.J.; Sijmonsma, T.P.; Broer, A.; Pfenninger, A.; Herzig, S.; Schmoll, D.; Broer, S. Mice lacking neutral amino acid transporter B(0)AT1 (Slc6a19) have elevated levels of FGF21 and GLP-1 and improved glycaemic control. Mol. Metab. 2015, 4, 406–417. [Google Scholar] [CrossRef]
- Javed, K.; Broer, S. Mice Lacking the Intestinal and Renal Neutral Amino Acid Transporter SLC6A19 Demonstrate the Relationship between Dietary Protein Intake and Amino Acid Malabsorption. Nutrients 2019, 11, 2024. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Shah, N.; Tiwari, P.K.; Javed, K.; Cheng, Q.; Aidhen, I.S.; Broer, S. Novel Chemical Scaffolds to Inhibit the Neutral Amino Acid Transporter B(0)AT1 (SLC6A19), a Potential Target to Treat Metabolic Diseases. Front. Pharmacol. 2020, 11, 140. [Google Scholar] [CrossRef]
- Thayer, T.C.; Wilson, S.B.; Mathews, C.E. Use of nonobese diabetic mice to understand human type 1 diabetes. Endocrinol. Metab. Clin. North. Am. 2010, 39, 541–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaglia, M.; Ahmed, S.; Anderson, M.S.; Atkinson, M.A.; Becker, D.; Bingley, P.J.; Bosi, E.; Brusko, T.M.; DiMeglio, L.A.; Evans-Molina, C.; et al. Introducing the Endotype Concept to Address the Challenge of Disease Heterogeneity in Type 1 Diabetes. Diabetes Care 2020, 43, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef]
- Cardinez, C.; Miraghazadeh, B.; Tanita, K.; da Silva, E.; Hoshino, A.; Okada, S.; Chand, R.; Asano, T.; Tsumura, M.; Yoshida, K.; et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J. Exp. Med. 2018, 215, 2715–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, D.A.; Fu, W.; Schonefeldt, S.; Feyerabend, T.B.; Ortiz-Lopez, A.; Lampi, Y.; Liston, A.; Mathis, D.; Rodewald, H.R. Type 1 diabetes in NOD mice unaffected by mast cell deficiency. Diabetes 2014, 63, 3827–3834. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waters, M.F.; Delghingaro-Augusto, V.; Javed, K.; Dahlstrom, J.E.; Burgio, G.; Bröer, S.; Nolan, C.J. Knockout of the Amino Acid Transporter SLC6A19 and Autoimmune Diabetes Incidence in Female Non-Obese Diabetic (NOD) Mice. Metabolites 2021, 11, 665. https://doi.org/10.3390/metabo11100665
Waters MF, Delghingaro-Augusto V, Javed K, Dahlstrom JE, Burgio G, Bröer S, Nolan CJ. Knockout of the Amino Acid Transporter SLC6A19 and Autoimmune Diabetes Incidence in Female Non-Obese Diabetic (NOD) Mice. Metabolites. 2021; 11(10):665. https://doi.org/10.3390/metabo11100665
Chicago/Turabian StyleWaters, Matthew F., Viviane Delghingaro-Augusto, Kiran Javed, Jane E. Dahlstrom, Gaetan Burgio, Stefan Bröer, and Christopher J. Nolan. 2021. "Knockout of the Amino Acid Transporter SLC6A19 and Autoimmune Diabetes Incidence in Female Non-Obese Diabetic (NOD) Mice" Metabolites 11, no. 10: 665. https://doi.org/10.3390/metabo11100665
APA StyleWaters, M. F., Delghingaro-Augusto, V., Javed, K., Dahlstrom, J. E., Burgio, G., Bröer, S., & Nolan, C. J. (2021). Knockout of the Amino Acid Transporter SLC6A19 and Autoimmune Diabetes Incidence in Female Non-Obese Diabetic (NOD) Mice. Metabolites, 11(10), 665. https://doi.org/10.3390/metabo11100665