
Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice

 , and
, and
Abstract
:
1. Introduction
2. Results
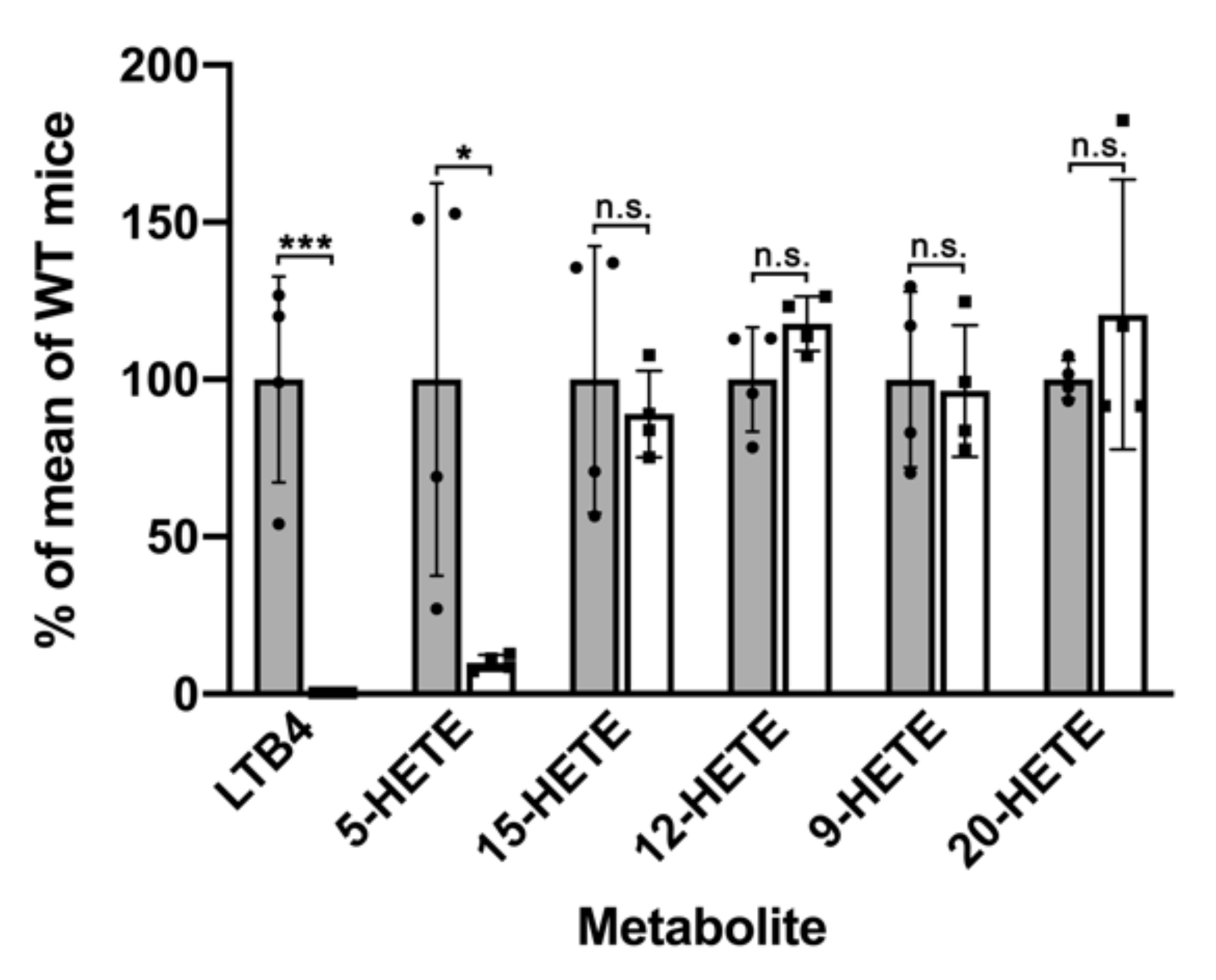
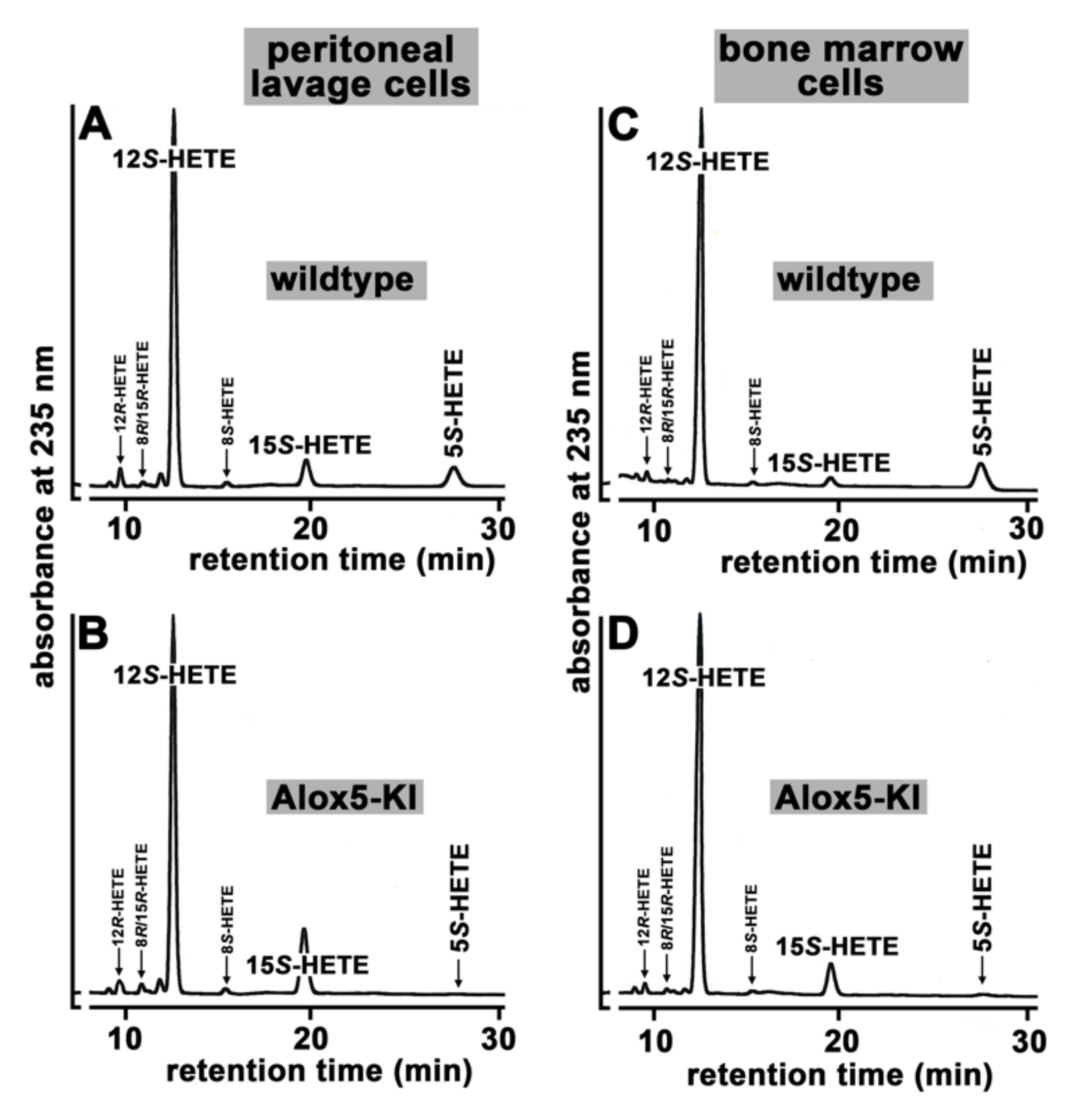
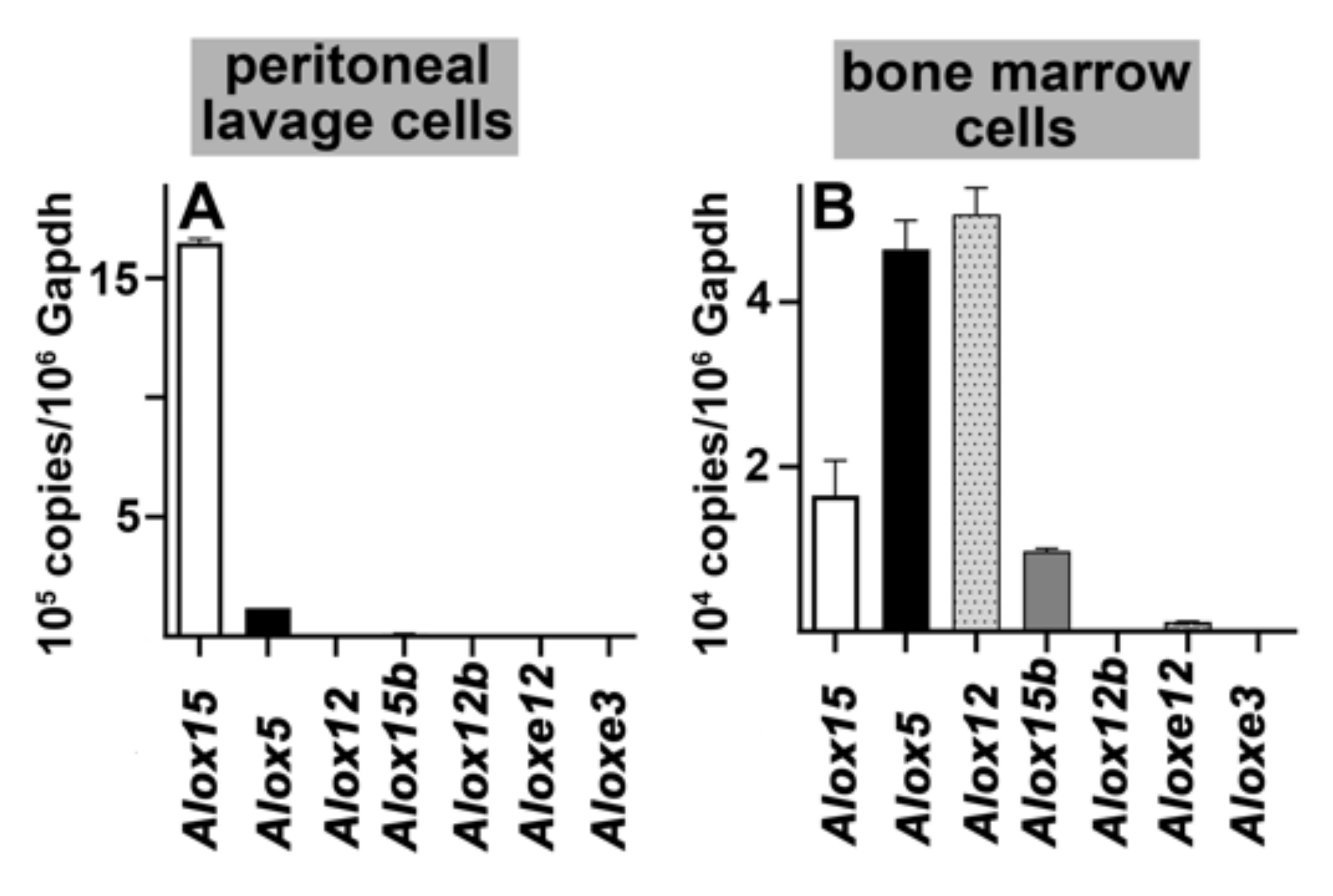
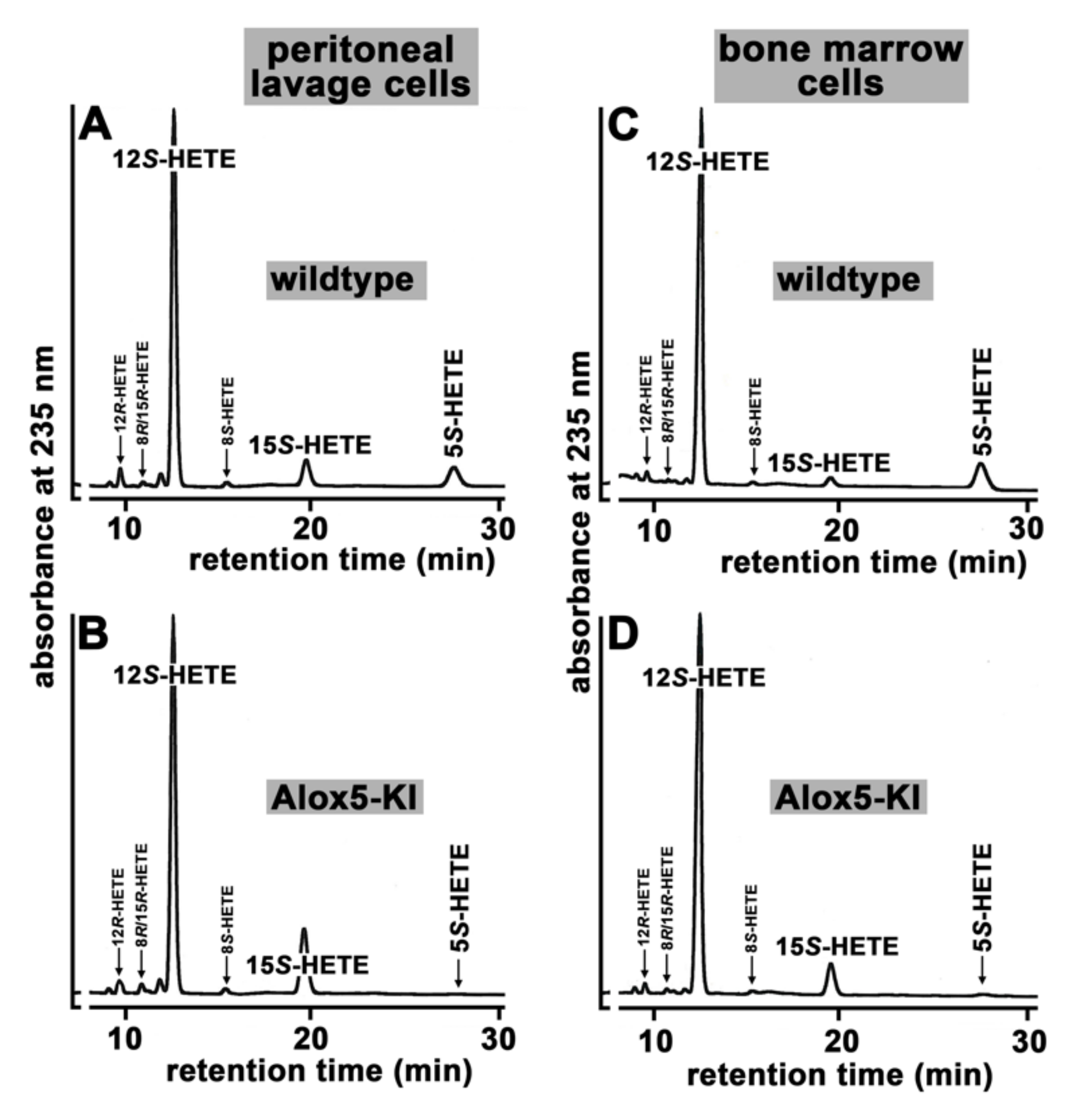
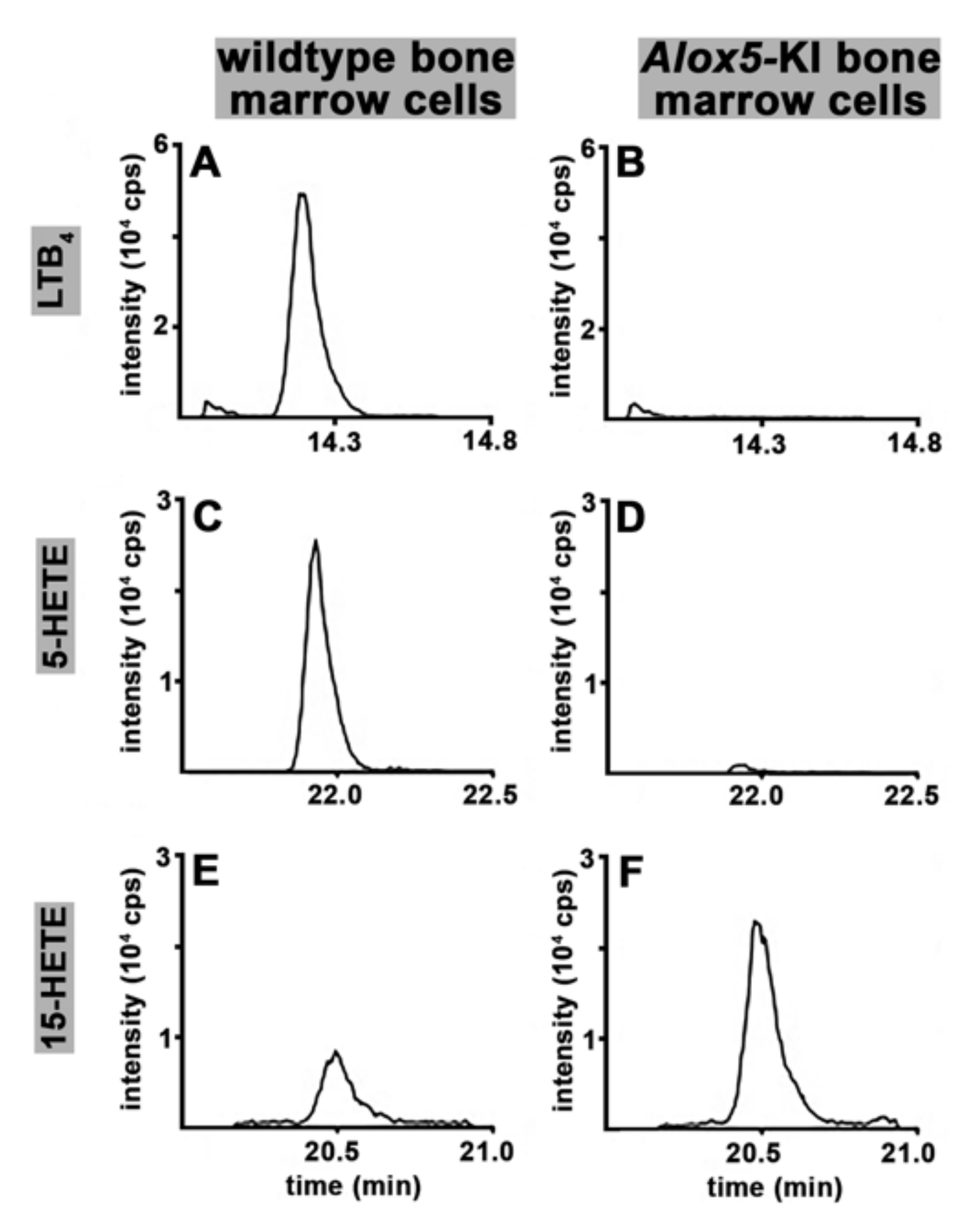
2.1. Functional Characterization of Alox5 Knock-in Mice (Alox5-KI)
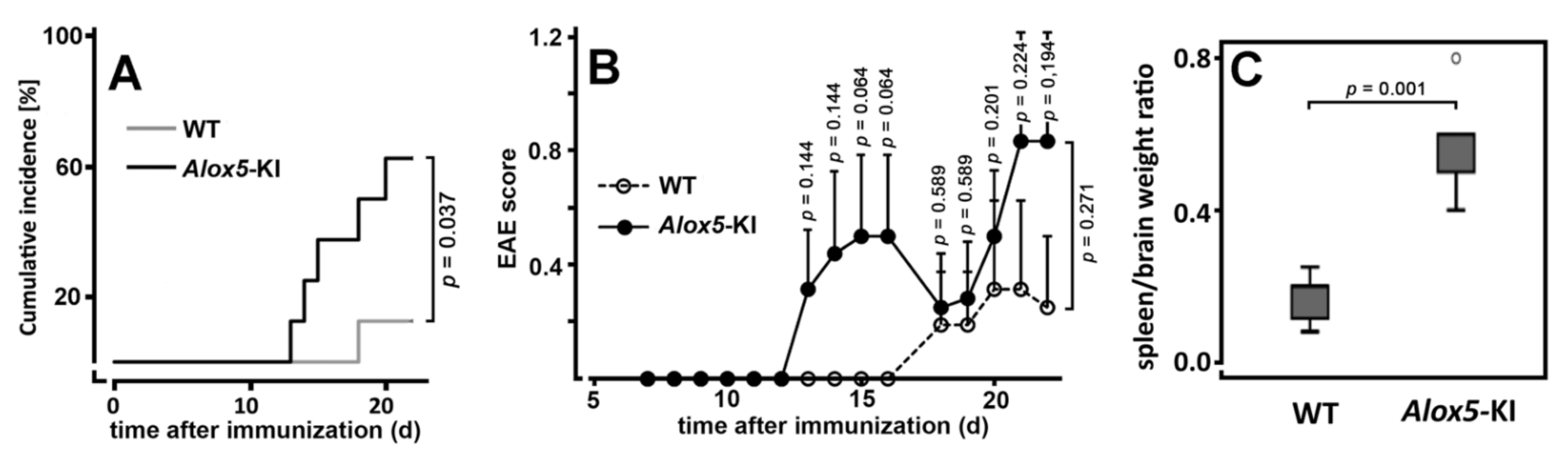
2.2. Alox5-KI Mice Show Earlier Disease Onset and Higher Cumulative Incidence Rates Than Wildtype Animals in the EAE Model
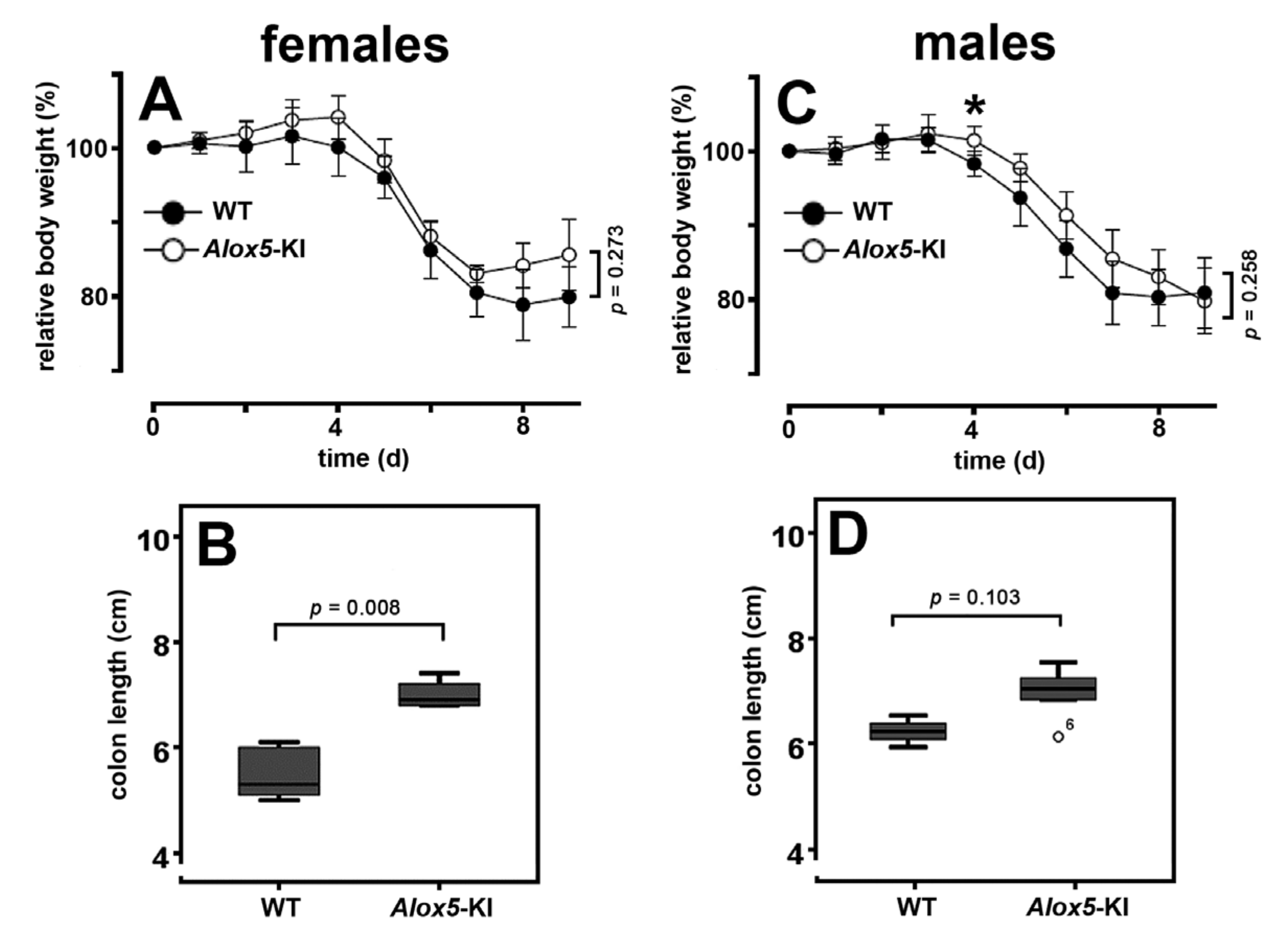
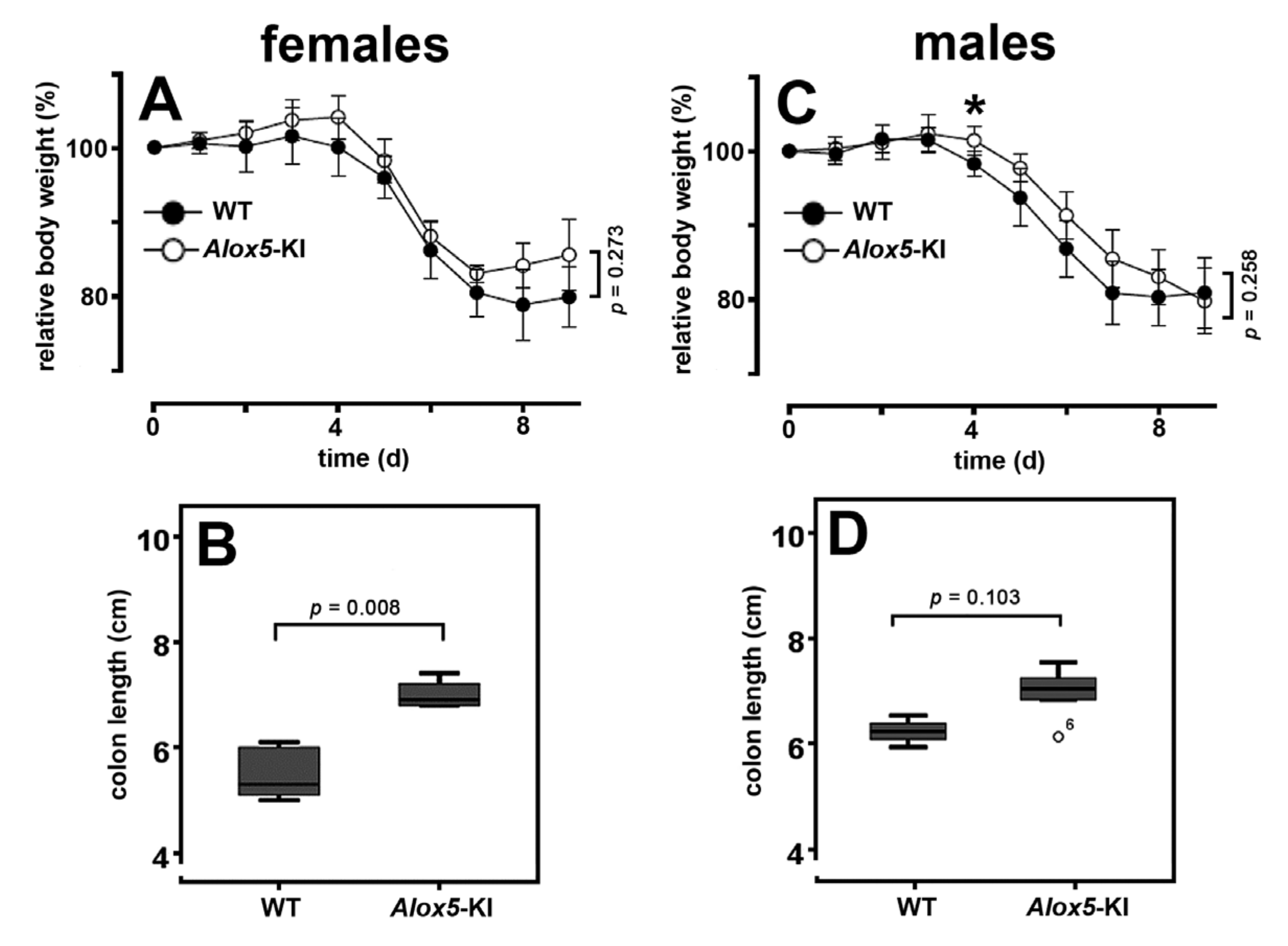
2.3. Alox5-KI Mice Are Not Protected from Inflammation in the DSS Colitis Model
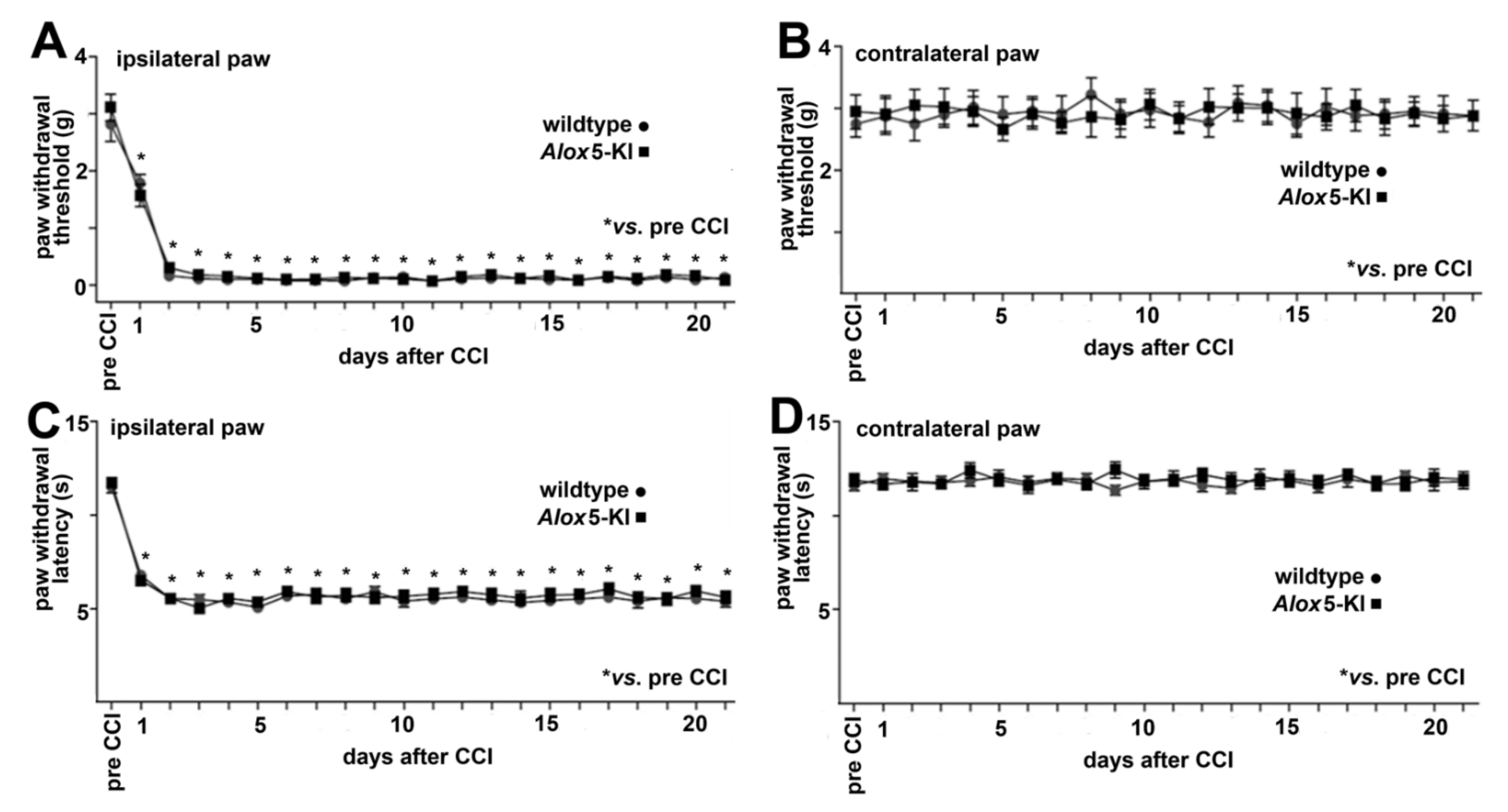
2.4. Alox5-KI Does Not Alter Mechanical and Heat Sensitivity in the Chronic Constriction Nerve Injury Model (CCI)
3. Discussion
3.1. Alox5-KI Mice Are Leukotriene Deficient but Exhibit an Increased Capacity for 15-HETE Formation
3.2. Alox5-KI Mice and Alox5−/− Animals Exhibit Differential Effects on EAE Score Kinetics
3.3. Alox5-KI Mice in the DSS Colitis Model
3.4. Alox5 and Neuropathic Pain
4. Materials and Methods
4.1. Chemicals
4.2. Genetically Modified Animals and Genotyping
4.3. Cell Preparation for Functional Characterization
4.4. RNA Extraction and qRT-PCR of the Different Alox-Isoforms
4.5. Cellular Ex Vivo Arachidonic Acid Oxygenase Activity Assays Using Exogenous Substrates
4.6. Ex Vivo Alox5 Activity Assays Using Heparinized Whole Peripheral Blood
4.7. Ex Vivo Formation of LTB4, 5-HETE and 15-HETE by Isolated Bone Marrow Cells Using Endogenous AA as Substrate
4.8. Experimental Autoimmune Encephalomyelitis (EAE) Model
4.9. Dextran Sodium Sulfate (DSS)-Induced Colitis Model
4.10. Chronic Constriction Injury (CCI) Model of Neuropathic Pain
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2015, 64, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, H.; Humeniuk, L.; Kozlov, N.; Roigas, S.; Adel, S.; Heydeck, D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog. Lipid Res. 2018, 72, 55–74. [Google Scholar] [CrossRef]
- Borgeat, P.; Hamberg, M.; Samuelsson, B. Transformation of arachidonic acid and homo-gamma-linolenic acid by rabbit polymorphonuclear leukocytes. Monohydroxy acids from novel lipoxygenases. J. Biol. Chem. 1976, 251, 7816–7820. [Google Scholar] [CrossRef]
- Morgan, E.L.; Maskrey, B.H.; Rowley, A.F. At what stage in metazoan evolution did leukotriene generation first appear?--key insights from cartilaginous fish. Dev. Comp. Immunol. 2005, 29, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Zou, Q.; Yu, T.; Song, C.; Huang, S.; Chen, S.; Ren, Z.; Xu, A. Ancestral genetic complexity of arachidonic acid metabolism in Metazoa. Biochim. Biophys. Acta 2014, 1841, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Tholander, F.; Muroya, A.; Roques, B.P.; Fournié-Zaluski, M.C.; Thunnissen, M.M.; Haeggström, J.Z. Structure-based dissection of the active site chemistry of leukotriene A4 hydrolase: Implications for M1 aminopeptidases and inhibitor design. Chem. Biol. 2008, 15, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Martinez Molina, D.; Wetterholm, A.; Kohl, A.; McCarthy, A.A.; Niegowski, D.; Ohlson, E.; Hammarberg, T.; Eshaghi, S.; Haeggström, J.Z.; Nordlund, P. Structural basis for synthesis of inflammatory mediators by human leukotriene C4 synthase. Nature 2007, 448, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Miyahara, N.; Gelfand, E.W. The role of leukotriene B(4) in allergic diseases. Allergol. Int. 2008, 57, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.J. Biological activities of leukotriene B4. Agents Actions 1981, 11, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Marom, Z.; Shelhamer, J.H.; Bach, M.K.; Morton, D.R.; Kaliner, M. Slow-reacting substances, leukotrienes C4 and D4, increase the release of mucus from human airways in vitro. Am. Rev. Respir. Dis. 1982, 126, 449–451. [Google Scholar] [PubMed]
- Matsuse, H.; Kohno, S. Leukotriene receptor antagonists pranlukast and montelukast for treating asthma. Expert Opin. Pharmacother. 2014, 15, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Berger, W.; De Chandt, M.T.; Cairns, C.B. Zileuton: Clinical implications of 5-Lipoxygenase inhibition in severe airway disease. Int. J. Clin. Pract. 2007, 61, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Sheller, J.R.; Johnson, E.N.; Funk, C.D. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature 1994, 372, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Moos, M.P.; Grabner, R.; Pedrono, F.; Fan, J.; Kaiser, B.; John, N.; Schmidt, S.; Spanbroek, R.; Lotzer, K.; et al. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat. Med. 2004, 10, 966–973. [Google Scholar] [CrossRef]
- Schewe, T.; Halangk, W.; Hiebsch, C.; Rapoport, S.M. A lipoxygenase in rabbit reticulocytes which attacks phospholipids and intact mitochondria. FEBS Lett. 1975, 60, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, S.M.; Schewe, T.; Wiesner, R.; Halangk, W.; Ludwig, P.; Janicke-Hohne, M.; Tannert, C.; Hiebsch, C.; Klatt, D. The lipoxygenase of reticulocytes. Purification, characterization and biological dynamics of the lipoxygenase; its identity with the respiratory inhibitors of the reticulocyte. Eur. J. Biochem. 1979, 96, 545–561. [Google Scholar] [CrossRef]
- Vogel, R.; Jansen, C.; Roffeis, J.; Reddanna, P.; Forsell, P.; Claesson, H.E.; Kuhn, H.; Walther, M. Applicability of the triad concept for the positional specificity of mammalian lipoxygenases. J. Biol. Chem. 2010, 285, 5369–5376. [Google Scholar] [CrossRef] [Green Version]
- Adel, S.; Karst, F.; Gonzalez-Lafont, A.; Pekarova, M.; Saura, P.; Masgrau, L.; Lluch, J.M.; Stehling, S.; Horn, T.; Kuhn, H.; et al. Evolutionary alteration of ALOX15 specificity optimizes the biosynthesis of antiinflammatory and proresolving lipoxins. Proc. Natl. Acad. Sci. USA 2016, 113, E4266–E4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, N.; Humeniuk, L.; Ufer, C.; Ivanov, I.; Golovanov, A.; Stehling, S.; Heydeck, D.; Kuhn, H. Functional characterization of novel ALOX15 orthologs representing key steps in mammalian evolution supports the Evolutionary Hypothesis of reaction specificity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1864, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.W.; Bailey, J.M.; Schewe, T.; Rapoport, S.M. Positional specificity of a reticulocyte lipoxygenase. Conversion of arachidonic acid to 15-S-hydroperoxy-eicosatetraenoic acid. J. Biol. Chem. 1982, 257, 6050–6055. [Google Scholar] [CrossRef]
- Kutzner, L.; Goloshchapova, K.; Heydeck, D.; Stehling, S.; Kuhn, H.; Schebb, N.H. Mammalian ALOX15 orthologs exhibit pronounced dual positional specificity with docosahexaenoic acid. Biochim. Biophys. Acta 2017, 1862, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Funk, C.D. Disruption of 12/15-lipoxygenase expression in peritoneal macrophages. Enhanced utilization of the 5-lipoxygenase pathway and diminished oxidation of low density lipoprotein. J. Biol. Chem. 1996, 271, 24055–24062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.R.; Heydeck, D.; Jones, G.W.; Krönke, G.; Funk, C.D.; Knapper, S.; Adams, D.; Kühn, H.; O’Donnell, V.B. Development of myeloproliferative disease in 12/15-lipoxygenase deficiency. Blood 2012, 119, 6173–6174. [Google Scholar] [CrossRef] [Green Version]
- Kronke, G.; Katzenbeisser, J.; Uderhardt, S.; Zaiss, M.M.; Scholtysek, C.; Schabbauer, G.; Zarbock, A.; Koenders, M.I.; Axmann, R.; Zwerina, J.; et al. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J. Immunol. 2009, 183, 3383–3389. [Google Scholar] [CrossRef]
- Kroschwald, S.; Chiu, C.Y.; Heydeck, D.; Rohwer, N.; Gehring, T.; Seifert, U.; Lux, A.; Rothe, M.; Weylandt, K.H.; Kuhn, H. Female mice carrying a defective Alox15 gene are protected from experimental colitis via sustained maintenance of the intestinal epithelial barrier function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 866–880. [Google Scholar] [CrossRef]
- Spite, M.; Claria, J.; Serhan, C.N. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014, 19, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Martin, H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat. Res. 2010, 690, 57–63. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.; Walther, M.; Anton, M.; Gerth, C.; Feussner, I.; Kuhn, H. Structural basis for lipoxygenase specificity. Conversion of the human leukocyte 5-lipoxygenase to a 15-lipoxygenating enzyme species by site-directed mutagenesis. J. Biol. Chem. 2001, 276, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofheinz, K.; Kakularam, K.R.; Adel, S.; Anton, M.; Polymarasetty, A.; Reddanna, P.; Kuhn, H.; Horn, T. Conversion of pro-inflammatory murine Alox5 into an anti-inflammatory 15S-lipoxygenating enzyme by multiple mutations of sequence determinants. Arch. Biochem. Biophys. 2013, 530, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Marbach-Breitrück, E.; Kutzner, L.; Rothe, M.; Gurke, R.; Schreiber, Y.; Reddanna, P.; Schebb, N.-H.; Stehling, S.; Wieler, L.; Heydeck, D.; et al. Functional charcterization of knock-in mice expressing a 12/15-lipoxygenating Alox5 mutant instead of the 5-lipoxygenating wild-type enzyme. Antioxid. Redox Signal. 2020, 32, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Emerson, M.R.; LeVine, S.M. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res. 2004, 1021, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Patil, C.S.; Kulkarni, S.K. Effect of 5-lipoxygenase inhibition on events associated with inflammatory bowel disease in rats. Indian J. Exp. Biol. 2004, 42, 667–673. [Google Scholar] [PubMed]
- Rund, K.M.; Ostermann, A.I.; Kutzner, L.; Galano, J.M.; Oger, C.; Vigor, C.; Wecklein, S.; Seiwert, N.; Durand, T.; Schebb, N.H. Development of an LC-ESI(-)-MS/MS method for the simultaneous quantification of 35 isoprostanes and isofurans derived from the major n3- and n6-PUFAs. Anal. Chim. Acta 2018, 1037, 63–74. [Google Scholar] [CrossRef]
- Steinhilber, D.; Hofmann, B. Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin. Pharmacol. Toxicol. 2014, 114, 70–77. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Wildfeuer, A.; Neu, I.S.; Safayhi, H.; Metzger, G.; Wehrmann, M.; Vogel, U.; Ammon, H.P. Effects of boswellic acids extracted from a herbal medicine on the biosynthesis of leukotrienes and the course of experimental autoimmune encephalomyelitis. Arzneim.-Forsch. 1998, 48, 668–674. [Google Scholar]
- Xu, J.; Zhang, Y.; Xiao, Y.; Ma, S.; Liu, Q.; Dang, S.; Jin, M.; Shi, Y.; Wan, B.; Zhang, Y. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARbeta/delta: A potential therapeutic role for CNS autoimmune disease. Cell Death Dis. 2013, 4, e569. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Hooper, K.M.; Ganea, D. The natural dual cyclooxygenase and 5-lipoxygenase inhibitor flavocoxid is protective in EAE through effects on Th1/Th17 differentiation and macrophage/microglia activation. Brain Behav. Immun. 2016, 53, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Whitney, L.W.; Ludwin, S.K.; McFarland, H.F.; Biddison, W.E. Microarray analysis of gene expression in multiple sclerosis and EAE identifies 5-lipoxygenase as a component of inflammatory lesions. J. Neuroimmunol. 2001, 121, 40–48. [Google Scholar] [CrossRef]
- DeVoss, J.; Diehl, L. Murine models of inflammatory bowel disease (IBD): Challenges of modeling human disease. Toxicol. Pathol. 2014, 42, 99–110. [Google Scholar] [CrossRef]
- Mazzon, E.; Sautebin, L.; Caputi, A.P.; Cuzzocrea, S. 5-lipoxygenase modulates the alteration of paracellular barrier function in mice ileum during experimental colitis. Shock 2006, 25, 377–383. [Google Scholar] [CrossRef]
- Davies, P.; Bailey, P.J.; Goldenberg, M.M.; Ford-Hutchinson, A.W. The role of arachidonic acid oxygenation products in pain and inflammation. Annu. Rev. Immunol. 1984, 2, 335–357. [Google Scholar] [CrossRef]
- Levine, J.D.; Lam, D.; Taiwo, Y.O.; Donatoni, P.; Goetzl, E.J. Hyperalgesic properties of 15-lipoxygenase products of arachidonic acid. Proc. Natl. Acad. Sci. USA 1986, 83, 5331–5334. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.W.; Cho, H.; Kwak, J.; Lee, S.Y.; Kang, C.J.; Jung, J.; Cho, S.; Min, K.H.; Suh, Y.G.; Kim, D.; et al. Direct activation of capsaicin receptors by products of lipoxygenases: Endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA 2000, 97, 6155–6160. [Google Scholar] [CrossRef] [Green Version]
- Lai, H.C.; Lu, C.H.; Wong, C.S.; Lin, B.F.; Chan, S.M.; Kuo, C.Y.; Wu, Z.F. Baicalein attenuates neuropathic pain and improves sciatic nerve function recovery in rats with partial sciatic nerve transection. J. Chin. Med. Assoc. 2018, 81, 955–963. [Google Scholar] [CrossRef]
- Aley, O.; Levine, J.D. Contribution of 5- and 12-lipoxygenase products to mechanical hyperalgesia induced by prostaglandin E(2) and epinephrine in the rat. Exp. Brain Res. 2003, 148, 482–487. [Google Scholar] [CrossRef]
- Singh, V.P.; Patil, C.S.; Kulkarni, S.K. Effect of zileuton in radicular pain induced by herniated nucleus pulposus in rats. Inflammopharmacology 2004, 12, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.G.; Holmin, S.; Mathiesen, T.; Meyerson, B.A.; Linderoth, B. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain 2000, 88, 239–248. [Google Scholar] [CrossRef]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Labuz, D.; Schmidt, Y.; Schreiter, A.; Rittner, H.L.; Mousa, S.A.; Machelska, H. Immune cell-derived opioids protect against neuropathic pain in mice. J. Clin. Investig. 2009, 119, 278–286. [Google Scholar] [CrossRef]
- Labuz, D.; Machelska, H. Stronger antinociceptive efficacy of opioids at the injured nerve trunk than at its peripheral terminals in neuropathic pain. J. Pharmacol. Exp. Ther. 2013, 346, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Adel, S.; Heydeck, D.; Kuhn, H.; Ufer, C. The lipoxygenase pathway in zebrafish. Expression and characterization of zebrafish ALOX5 and comparison with its human ortholog. Biochim. Biophys. Acta 2016, 1861, 1–11. [Google Scholar] [CrossRef]
- Gladue, R.P.; Carroll, L.A.; Milici, A.J.; Scampoli, D.N.; Stukenbrok, H.A.; Pettipher, E.R.; Salter, E.D.; Contillo, L.; Showell, H.J. Inhibition of leukotriene B4-receptor interaction suppresses eosinophil infiltration and disease pathology in a murine model of experimental allergic encephalomyelitis. J. Exp. Med. 1996, 183, 1893–1898. [Google Scholar] [CrossRef] [Green Version]
- Simmons, R.D.; Hugh, A.R.; Willenborg, D.O.; Cowden, W.B. Suppression of active but not passive autoimmune encephalomyelitis by dual cyclo-oxygenase and 5-lipoxygenase inhibition. Acta Neurol. Scand. 1992, 85, 197–199. [Google Scholar] [CrossRef]
- Limor, R.; Sharon, O.; Knoll, E.; Many, A.; Weisinger, G.; Stern, N. Lipoxygenase-derived metabolites are regulators of peroxisome proliferator-activated receptor gamma-2 expression in human vascular smooth muscle cells. Am. J. Hypertens. 2008, 21, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Wu, Y.; Morris, J.S.; Stimmel, J.B.; Leesnitzer, L.M.; Fischer, S.M.; Lippman, S.M.; Shureiqi, I. Oxidative metabolism of linoleic acid modulates PPAR-beta/delta suppression of PPAR-gamma activity. Oncogene 2006, 25, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Pallio, G.; Bitto, A.; Pizzino, G.; Galfo, F.; Irrera, N.; Minutoli, L.; Arcoraci, V.; Squadrito, G.; Macri, A.; Squadrito, F.; et al. Use of a balanced dual cyclooxygenase-1/2 and 5-lypoxygenase inhibitor in experimental colitis. Eur. J. Pharmacol. 2016, 789, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Labuz, D.; Celik, M.Ö.; Zimmer, A.; Machelska, H. Distinct roles of exogenous opioid agonists and endogenous opioid peptides in the peripheral control of neuropathy-triggered heat pain. Sci. Rep. 2016, 6, 32799. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 1, 77–88. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Genotype | Product (%) | ||
|---|---|---|---|---|
| 5S-HETE | 12S-HETE | 15S-HETE | ||
| Peritoneal lavage cells | WT | 7.8 ± 2.7 | 84.1 ± 2.9 | 8.1 ± 0.4 |
| Alox5-KI | 0 * | 82.4 ± 2.4 # | 18.0 ± 2.4 * | |
| Bone marrow cells | WT | 12.0 ± 1.4 | 85.8 ± 1.8 | 2.2 ± 0.5 |
| Alox5-KI | 0 * | 89.7 ± 0.4 * | 10.3 ± 0.4 * | |
| Gene | Forward Primer 5′→ 3′ | Reverse Primer 5′ → 3′ |
|---|---|---|
| mGapdh | CCATCACCATCTTCCAGAGCGA | GGATGACCTTGCCCACAGCCTTG |
| mAlox15 | GTACGCGGGCTCCAACAACGA | TCTCCGGGGCCCTTCACAGAA |
| mAlox15b | CCTCCCGCTTATGTCTTTCCGT | GCCCTTTGACTTTCAGCTCCGTA |
| mAlox12 | GCGGCCATGTTCAGTTGCTTAC | CATCGTCACGTCGTCCTTGCTG |
| mAlox5 | TCGAGTTCCCATGTTACCGCT | CTGTGGTCACTGGGAGCTTCG |
| mAlox12b | GGTGATGGTTCGGGGTCTGTCT | GAGTCCAGAGCACCAAGAGCACA |
| mAloxe12 | CTCCAGCCACCACGACACGG | GCAACGAGTCCACAATGTCCCT |
| mAloxe3 | GGGCGGCTATTGAGAGGTTTGT | TCTGGTCCTTTGGCTCTTGGCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marbach-Breitrück, E.; Rohwer, N.; Infante-Duarte, C.; Romero-Suarez, S.; Labuz, D.; Machelska, H.; Kutzner, L.; Schebb, N.H.; Rothe, M.; Reddanna, P.; et al. Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice. Metabolites 2021, 11, 698. https://doi.org/10.3390/metabo11100698
Marbach-Breitrück E, Rohwer N, Infante-Duarte C, Romero-Suarez S, Labuz D, Machelska H, Kutzner L, Schebb NH, Rothe M, Reddanna P, et al. Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice. Metabolites. 2021; 11(10):698. https://doi.org/10.3390/metabo11100698
Chicago/Turabian StyleMarbach-Breitrück, Eugenia, Nadine Rohwer, Carmen Infante-Duarte, Silvina Romero-Suarez, Dominika Labuz, Halina Machelska, Laura Kutzner, Nils Helge Schebb, Michael Rothe, Pallu Reddanna, and et al. 2021. "Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice" Metabolites 11, no. 10: 698. https://doi.org/10.3390/metabo11100698
APA StyleMarbach-Breitrück, E., Rohwer, N., Infante-Duarte, C., Romero-Suarez, S., Labuz, D., Machelska, H., Kutzner, L., Schebb, N. H., Rothe, M., Reddanna, P., Weylandt, K. H., Wieler, L. H., Heydeck, D., & Kuhn, H. (2021). Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice. Metabolites, 11(10), 698. https://doi.org/10.3390/metabo11100698