
Deep Learning for Rapid Identification of Microbes Using Metabolomics Profiles




Abstract
:
1. Introduction
2. Results
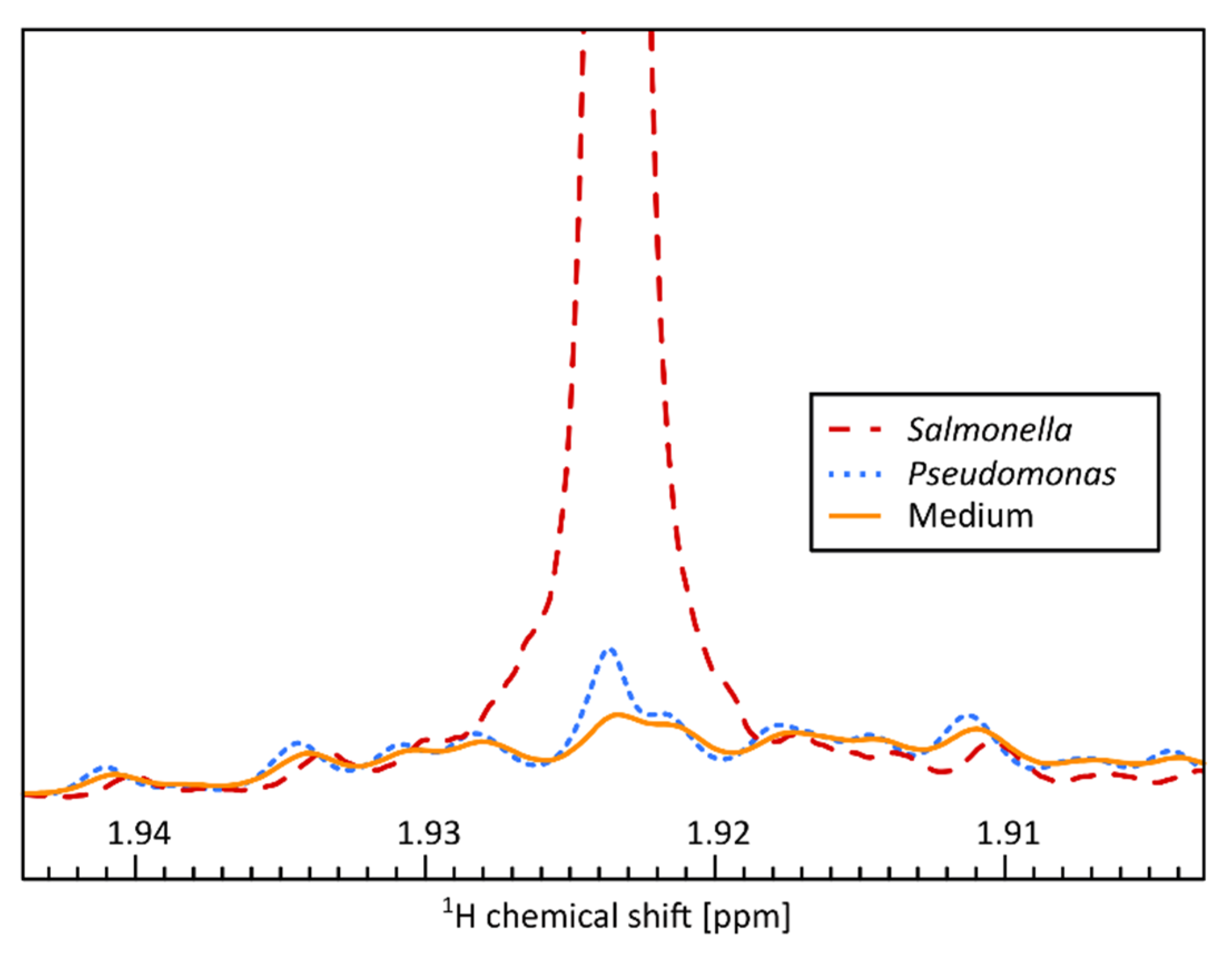
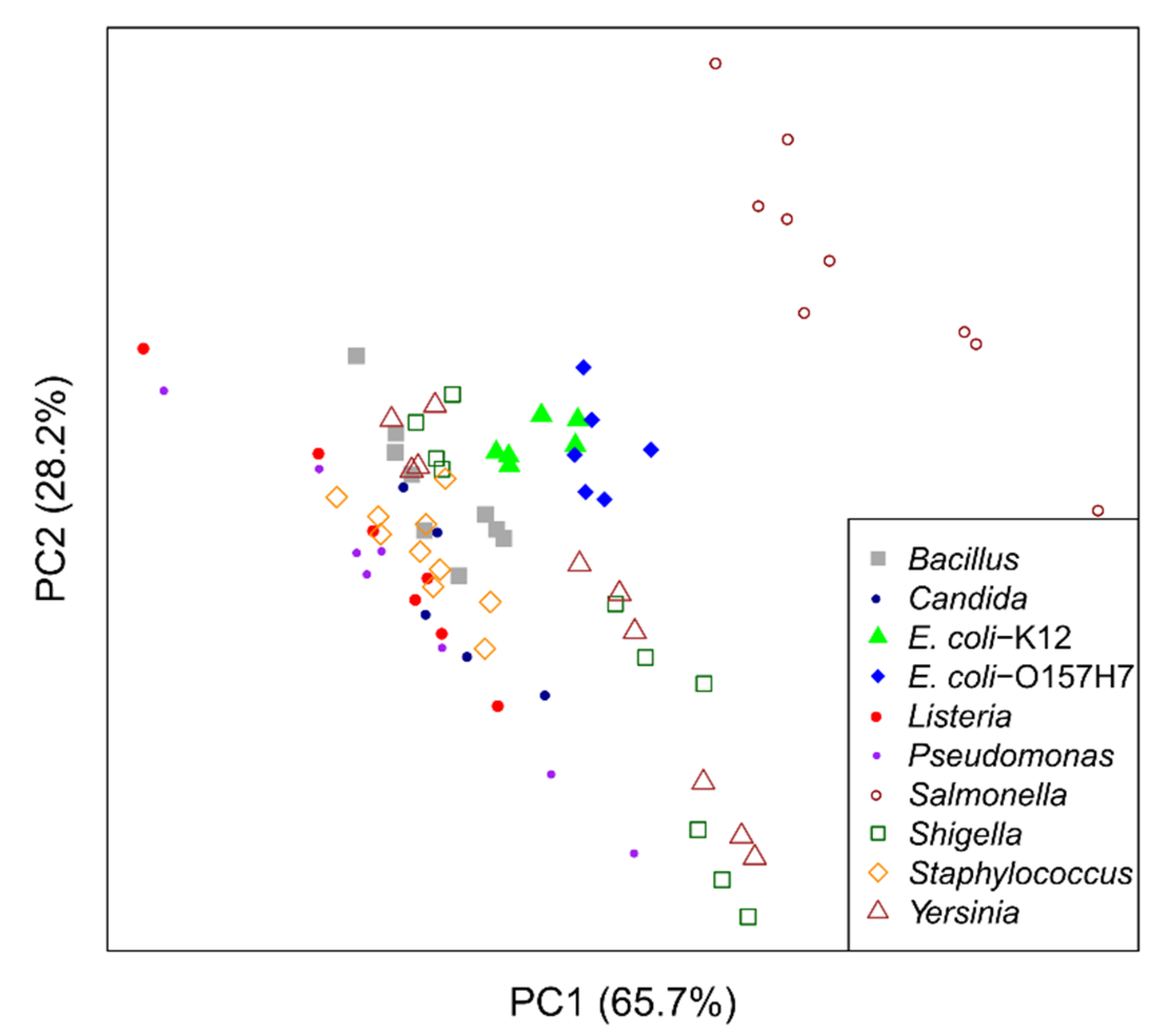
2.1. Unsupervised Approach
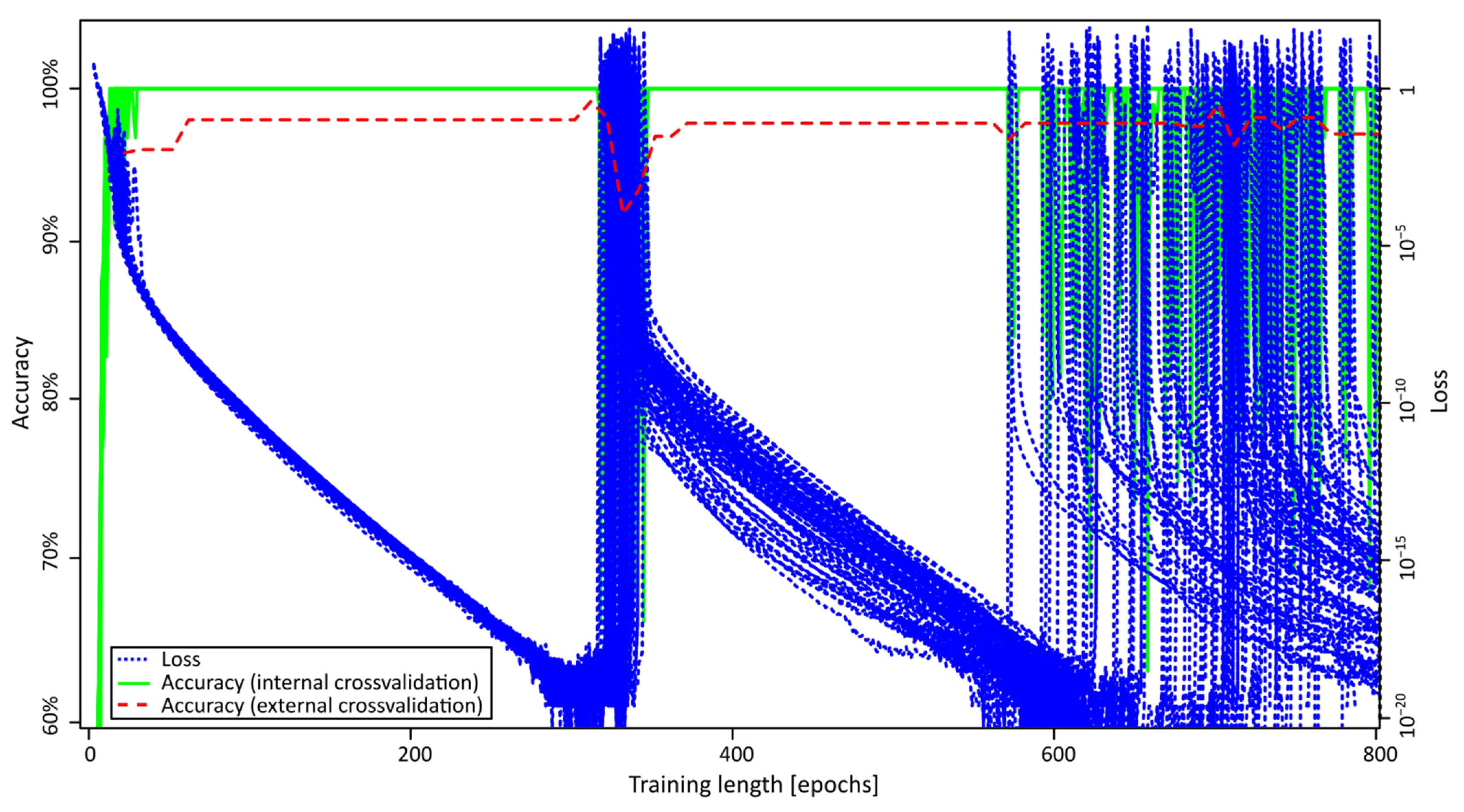
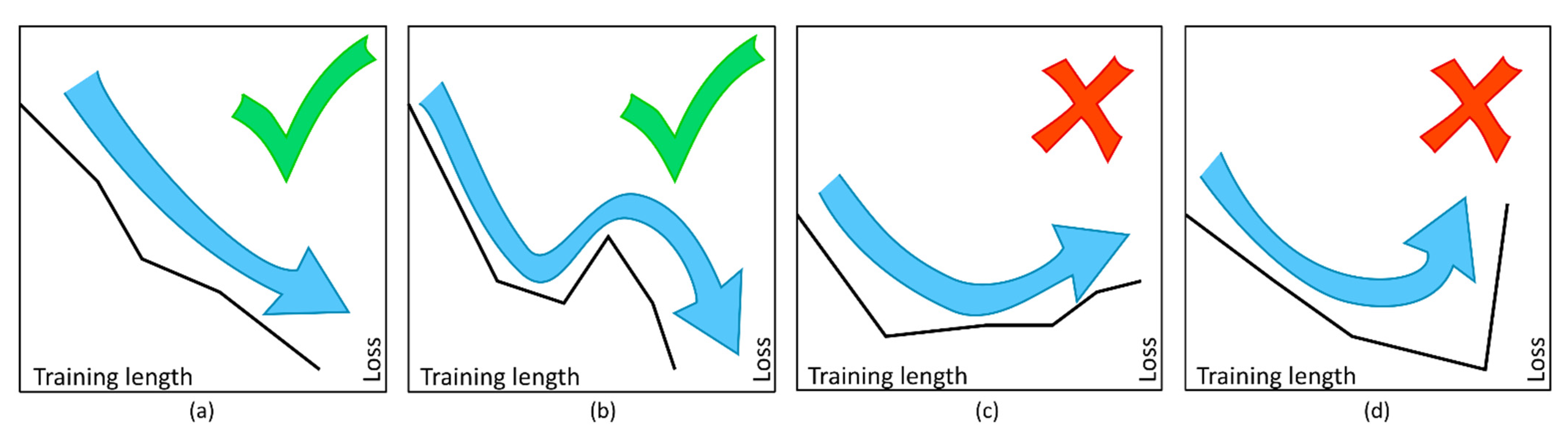
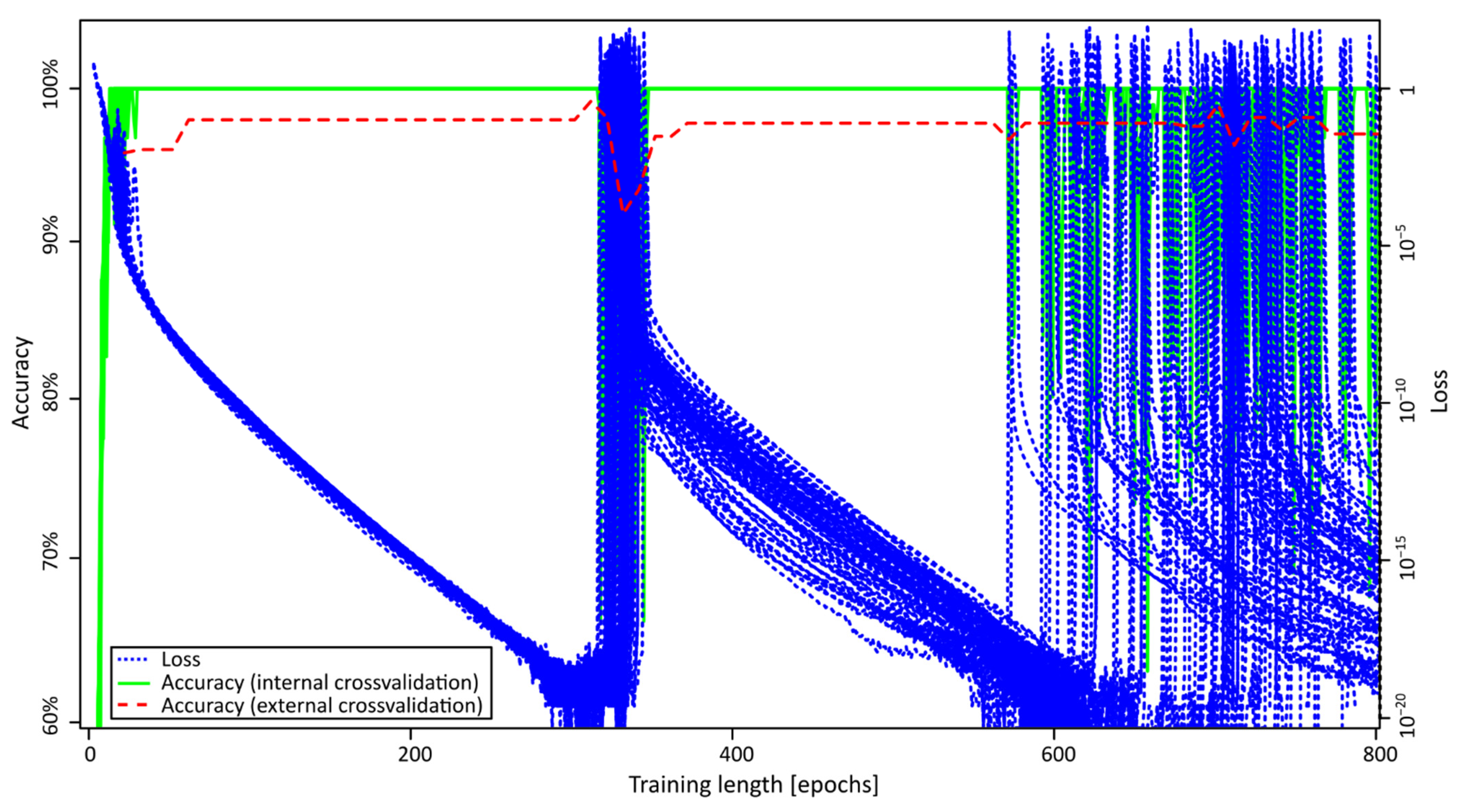
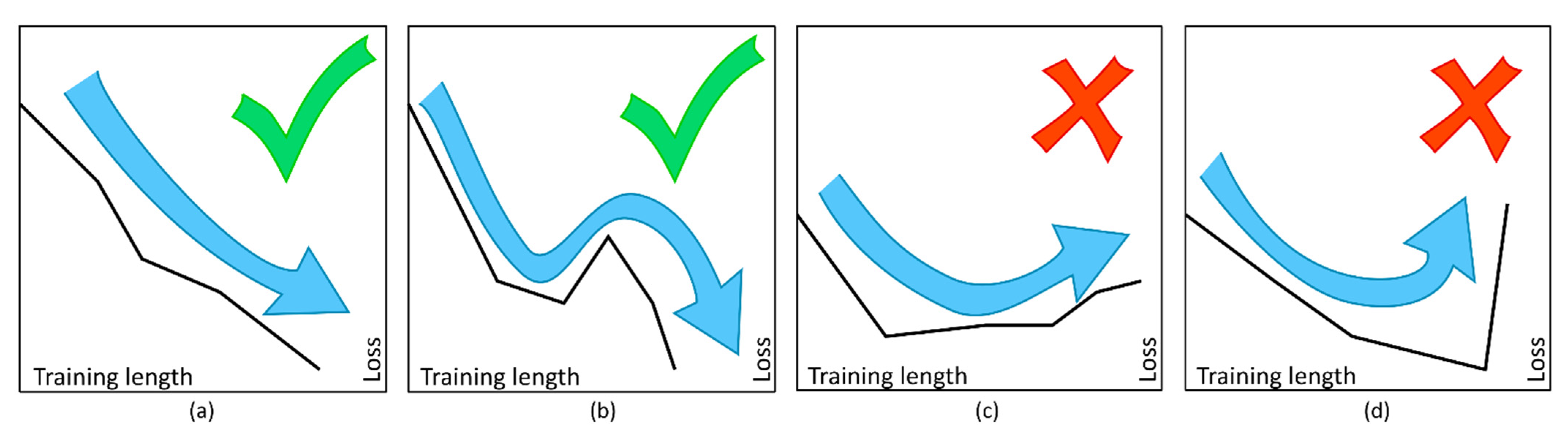
2.2. ANN Training
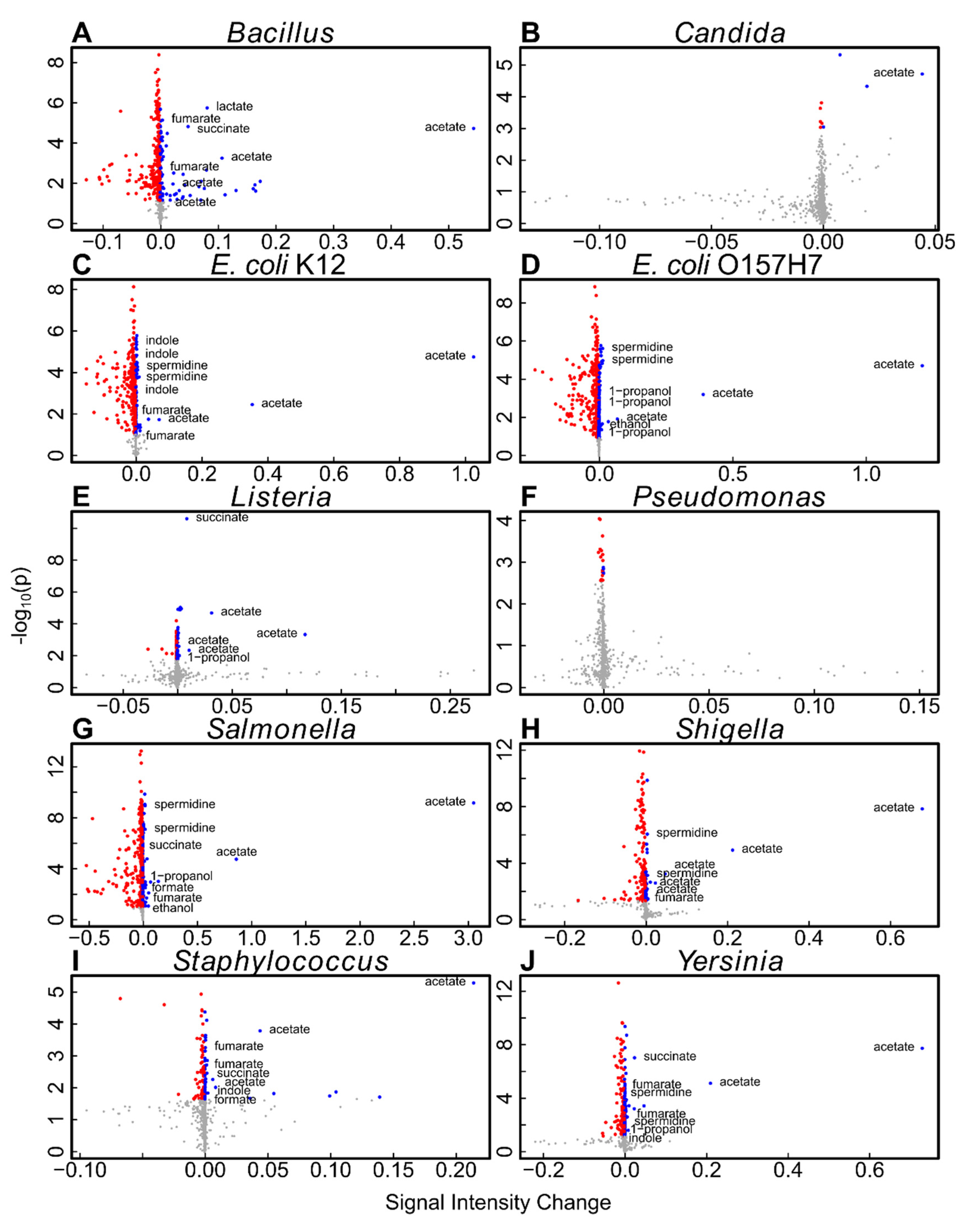
2.3. Method Comparison
3. Discussion
4. Materials and Methods
4.1. Microorganism Cultivation
4.2. NMR Measurements
4.3. Data Preparation
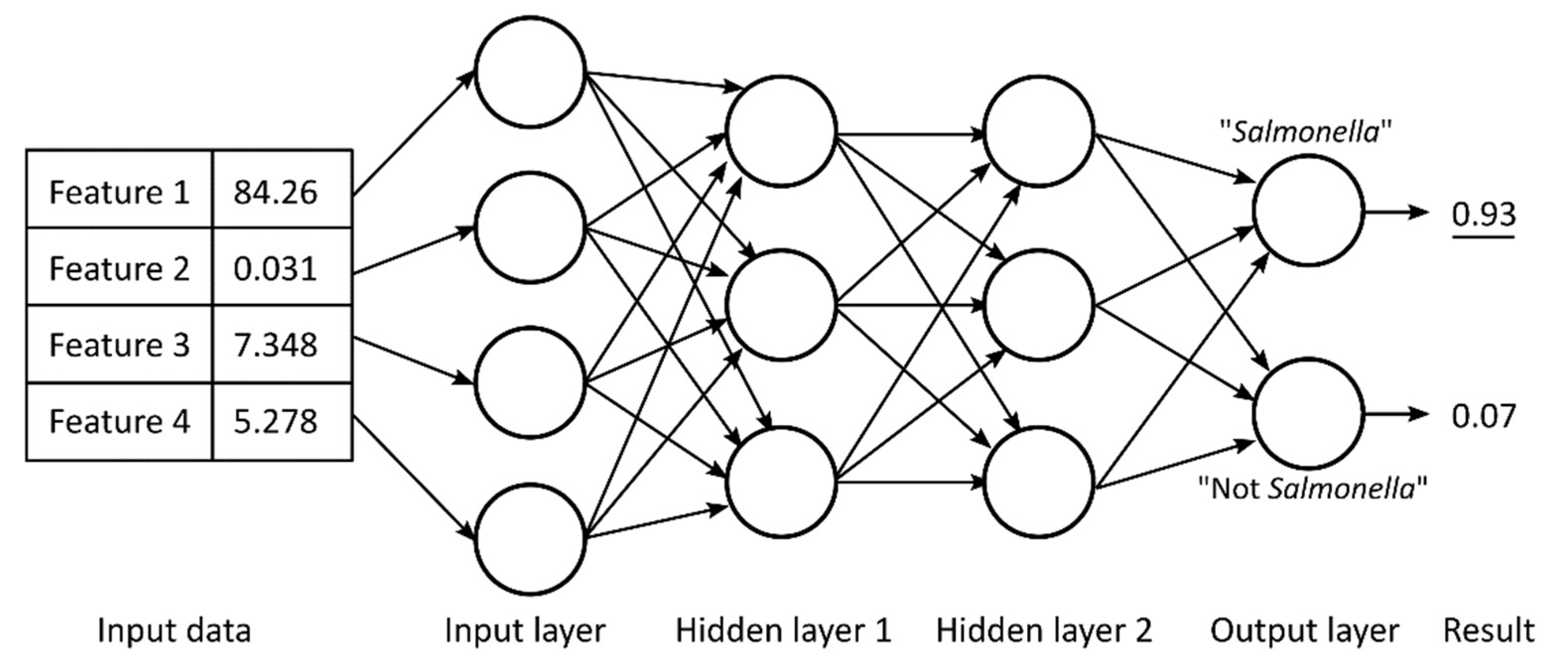
4.4. Classification
4.5. Statistical Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Estimates of the Global Burden of Foodborne Diseases: Foodborne Disease Burden Epidemiology Reference Group 2007–2015; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Charlebois, S.; Juhasz, M.; Music, J.; Vézeau, J. A review of Canadian and international food safety systems: Issues and recommendations for the future. Compr. Rev. Food Sci. Food Saf. 2021, 20, 5043–5066. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.R.; Shah, R.M.; Karpe, A.V.; Morrison, P.D.; Kouremenos, K.; Beale, D.J.; Palombo, E.A. Detection of Foodborne Pathogens Using Proteomics and Metabolomics-Based Approaches. Front. Microbiol. 2018, 9, 3132. [Google Scholar] [CrossRef]
- Rautureau, G.J.P.; Palama, T.L.; Canard, I.; Mirande, C.; Chatellier, S.; van Belkum, A.; Elena-Herrmann, B. Discrimination of Escherichia coli and Shigella spp. by Nuclear Magnetic Resonance Based Metabolomic Characterization of Culture Media. ACS Infect. Dis. 2019, 5, 1879–1886. [Google Scholar] [CrossRef]
- Kriegeskorte, N. Deep Neural Networks: A New Framework for Modeling Biological Vision and Brain Information Processing. Annu. Rev. Vis. Sci. 2015, 1, 417–446. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Lamichhane, S.; Mathema, V.B.; McGlinchey, A.; Dickens, A.M.; Khoomrung, S.; Orešič, M. Deep learning meets metabolomics: A methodological perspective. Brief. Bioinform. 2021, 22, 1531–1542. [Google Scholar] [CrossRef]
- Pomyen, Y.; Wanichthanarak, K.; Poungsombat, P.; Fahrmann, J.; Grapov, D.; Khoomrung, S. Deep metabolome: Applications of deep learning in metabolomics. Comput. Struct. Biotechnol. J. 2020, 18, 2818–2825. [Google Scholar] [CrossRef]
- Fukushima, K. Visual Feature Extraction by a Multilayered Network of Analog Threshold Elements. IEEE Trans. Syst. Sci. Cybern. 1969, 5, 322–333. [Google Scholar] [CrossRef]
- Diederik, P.; Kingma, J.L.B. Adam: A Method for Stochastic Optimization. In Proceedings of the International Conference on Learning Representations (ICLR), San Diego, CA, USA, 7–9 May 2015; Available online: https://hdl.handle.net/11245/1.505367 (accessed on 30 November 2021).
- Wang, D.; Greenwood, P.; Klein, M.S. A Protein-free Chemically Defined Medium for the Cultivation of Various Microorganisms with Food Safety Significance. J. Appl. Microbiol. 2021, 131, 844–854. [Google Scholar] [CrossRef]
- Hochrein, J.; Klein, M.S.; Zacharias, H.U.; Li, J.; Wijffels, G.; Schirra, H.J.; Spang, R.; Oefner, P.J.; Gronwald, W. Performance evaluation of algorithms for the classification of metabolic 1H NMR fingerprints. J. Proteome Res. 2012, 11, 6242–6251. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, E.; Duport, C.; Zigha, A.; Schmitt, P. Characterization of aerobic and anaerobic vegetative growth of the food-borne pathogen Bacillus cereus F4430/73 strain. Can. J. Microbiol. 2005, 51, 149–158. [Google Scholar] [CrossRef]
- Willetts, A. Ester formation from ethanol by Candida pseudotropicalis. Antonie Leeuwenhoek 1989, 56, 175–180. [Google Scholar] [CrossRef]
- Landwall, P.; Holme, T. Influence of Glucose and Dissolved Oxygen Concentrations on Yields of Escherichia colib in Dialysis Culture. Microbiology 1977, 103, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Romick, T.L.; Fleming, H.P.; McFeeters, R.F. Aerobic and anaerobic metabolism of Listeria monocytogenes in defined glucose medium. Appl. Environ. Microbiol. 1996, 62, 304–307. [Google Scholar] [CrossRef] [Green Version]
- Huseby, D.L.; Roth, J.R. Evidence that a metabolic microcompartment contains and recycles private cofactor pools. J. Bacteriol. 2013, 195, 2864–2879. [Google Scholar] [CrossRef] [Green Version]
- Kentner, D.; Martano, G.; Callon, M.; Chiquet, P.; Brodmann, M.; Burton, O.; Wahlander, A.; Nanni, P.; Delmotte, N.; Grossmann, J.; et al. Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc. Natl. Acad. Sci. USA 2014, 111, 9929–9934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halsey Cortney, R.; Lei, S.; Wax Jacqueline, K.; Lehman Mckenzie, K.; Nuxoll Austin, S.; Steinke, L.; Sadykov, M.; Powers, R.; Fey Paul, D.; Richardson Anthony, R.; et al. Amino Acid Catabolism in Staphylococcus aureus and the Function of Carbon Catabolite Repression. mBio 2017, 8, e01434-16. [Google Scholar] [CrossRef] [Green Version]
- Bücker, R.; Heroven, A.K.; Becker, J.; Dersch, P.; Wittmann, C. The pyruvate-tricarboxylic acid cycle node: A focal point of virulence control in the enteric pathogen Yersinia pseudotuberculosis. J. Biol. Chem. 2014, 289, 30114–30132. [Google Scholar] [CrossRef] [Green Version]
- Boumba, V.A.; Kourkoumelis, N.; Gousia, P.; Economou, V.; Papadopoulou, C.; Vougiouklakis, T. Modeling microbial ethanol production by E. coli under aerobic/anaerobic conditions: Applicability to real postmortem cases and to postmortem blood derived microbial cultures. Forensic Sci. Int. 2013, 232, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Sampson Edith, M.; Bobik Thomas, A. Microcompartments for B12-Dependent 1,2-Propanediol Degradation Provide Protection from DNA and Cellular Damage by a Reactive Metabolic Intermediate. J. Bacteriol. 2008, 190, 2966–2971. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Murrieta Charles, M.; Rule Daniel, C.; Miller Kurt, W. Exogenous or l-Rhamnose-Derived 1,2-Propanediol Is Metabolized via a pduD-Dependent Pathway in Listeria innocua. Appl. Environ. Microbiol. 2008, 74, 7073–7079. [Google Scholar] [CrossRef] [Green Version]
- Sargo, C.R.; Campani, G.; Silva, G.G.; Giordano, R.C.; Da Silva, A.J.; Zangirolami, T.C.; Correia, D.M.; Ferreira, E.C.; Rocha, I. Salmonella typhimurium and Escherichia coli dissimilarity: Closely related bacteria with distinct metabolic profiles. Biotechnol. Prog. 2015, 31, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-L.; Zhang, S.-K.; Chen, J.-Y.; Han, B.-Z. Metabolic profiling of Staphylococcus aureus cultivated under aerobic and anaerobic conditions with 1H NMR-based nontargeted analysis. Can. J. Microbiol. 2012, 58, 709–718. [Google Scholar] [CrossRef]
- Strecker, A.; Schubert, C.; Zedler, S.; Steinmetz, P.; Unden, G. DcuA of aerobically grown Escherichia coli serves as a nitrogen shuttle (L-aspartate/fumarate) for nitrogen uptake. Mol. Microbiol. 2018, 109, 801–811. [Google Scholar] [CrossRef]
- Han, T.H.; Lee, J.-H.; Cho, M.H.; Wood, T.K.; Lee, J. Environmental factors affecting indole production in Escherichia coli. Res. Microbiol. 2011, 162, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, H.; Yahata, M. l-Lactic acid production by Bacillus sp. in anaerobic and aerobic culture. J. Ferment. Bioeng. 1996, 81, 272–274. [Google Scholar] [CrossRef]
- Pugin, B.; Barcik, W.; Westermann, P.; Heider, A.; Wawrzyniak, M.; Hellings, P.; Akdis, C.A.; O’Mahony, L. A wide diversity of bacteria from the human gut produces and degrades biogenic amines. Microb. Ecol. Health Dis. 2017, 28, 1353881. [Google Scholar] [CrossRef]
- Feehily, C.; O’Byrne, C.P.; Karatzas, K.A.G. Functional γ-Aminobutyrate Shunt in Listeria monocytogenes: Role in acid tolerance and succinate biosynthesis. Appl. Environ. Microbiol. 2013, 79, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.S. Affine Transformation of Negative Values for NMR Metabolomics Using the mrbin R Package. J. Proteome Res. 2021, 20, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
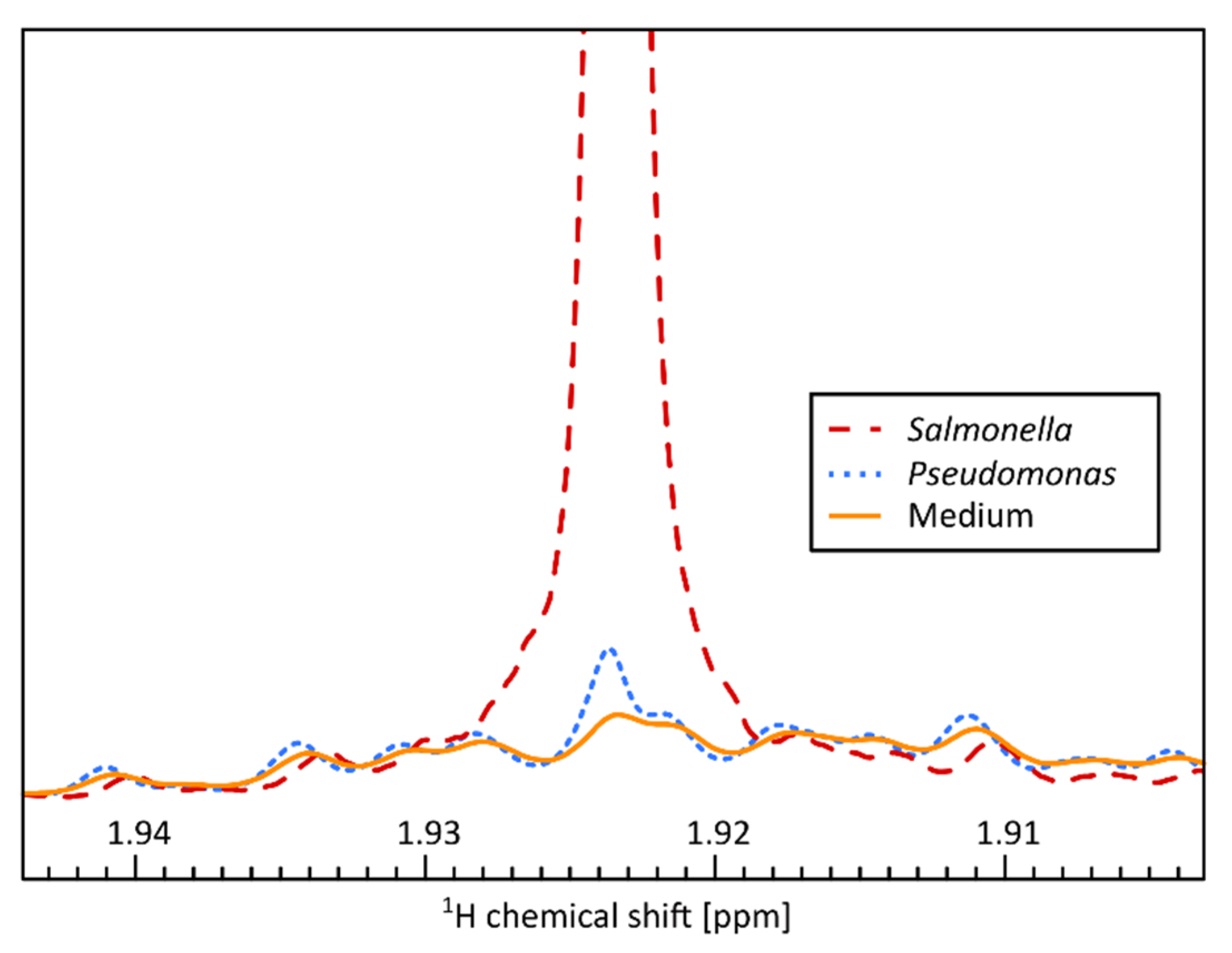{kind=link}
| Accuracy 1 | ||
|---|---|---|
| Method | Using All Signals | Using only Increasing Signals |
| Artificial neural networks (ANN) | 91.2% ± 1.5% | 99.2% ± 1.0% |
| Random forests (RF) | 89.8% ± 2.2% | 96.5% ± 1.6% |
| Support vector machines (SVM) | 88.8% ± 0.0% | 94.6% ± 0.0% |
| True | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacillus | Candida | E. coli-K12 | E. coli-O157H7 | Listeria | Pseudomonas | Salmonella | Shigella | Staphylococcus | Yersinia | ||
| predicted | Bacillus | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| Candida | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | |
| E. coli-K12 | 0% | 0% | 100% | 0% | 0% | 0% | 0% | 3.3% | 0% | 0% | |
| E. coli-O157H7 | 0% | 0% | 0% | 100% | 0% | 0% | 0% | 0% | 0% | 0% | |
| Listeria | 0% | 0% | 0% | 0% | 95.2% | 0% | 0% | 0% | 0% | 0% | |
| Pseudomonas | 0% | 0% | 0% | 0% | 4.8% | 100% | 0% | 0% | 0% | 0% | |
| Salmonella | 0% | 0% | 0% | 0% | 0% | 0% | 100% | 0% | 0% | 0% | |
| Shigella | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 96.7% | 0% | 0% | |
| Staphylococcus | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 100% | 0% | |
| Yersinia | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 100% | |
| Bacillus | Candida | E. coli-K12 | E. coli-O157H7 | Listeria | Pseudomonas | Salmonella | Shigella | Staphylococcus | Yersinia | |
|---|---|---|---|---|---|---|---|---|---|---|
| Acetic acid | + | + | + | + | + | 0 | + | + | + | + |
| Ethanol | 0 | 0 | 0 | + | 0 | 0 | + | 0 | 0 | 0 |
| Formic acid | 0 | 0 | 0 | 0 | 0 | 0 | + | 0 | + | 0 |
| Fumaric acid | + | 0 | + | 0 | 0 | 0 | + | + | + | + |
| Indole | 0 | 0 | + | 0 | 0 | 0 | 0 | 0 | + | + |
| Lactic acid | + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1-Propanol | 0 | 0 | 0 | + | + | 0 | + | 0 | 0 | + |
| Spermidine | 0 | 0 | + | + | 0 | 0 | + | + | 0 | + |
| Succinic acid | + | 0 | 0 | 0 | + | 0 | + | 0 | 0 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Greenwood, P.; Klein, M.S. Deep Learning for Rapid Identification of Microbes Using Metabolomics Profiles. Metabolites 2021, 11, 863. https://doi.org/10.3390/metabo11120863
Wang D, Greenwood P, Klein MS. Deep Learning for Rapid Identification of Microbes Using Metabolomics Profiles. Metabolites. 2021; 11(12):863. https://doi.org/10.3390/metabo11120863
Chicago/Turabian StyleWang, Danhui, Peyton Greenwood, and Matthias S. Klein. 2021. "Deep Learning for Rapid Identification of Microbes Using Metabolomics Profiles" Metabolites 11, no. 12: 863. https://doi.org/10.3390/metabo11120863
APA StyleWang, D., Greenwood, P., & Klein, M. S. (2021). Deep Learning for Rapid Identification of Microbes Using Metabolomics Profiles. Metabolites, 11(12), 863. https://doi.org/10.3390/metabo11120863