
Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
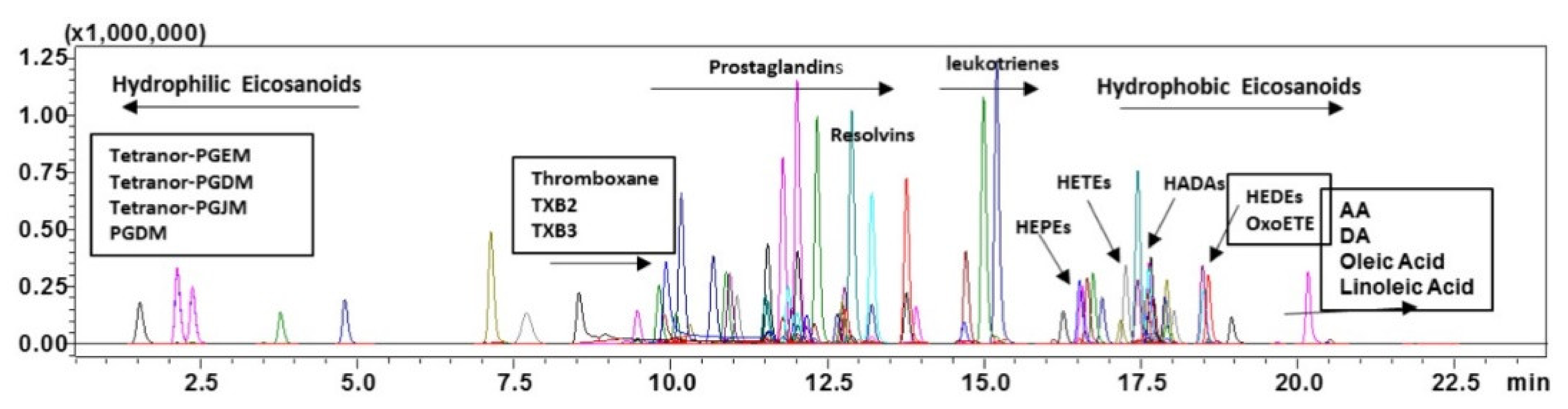
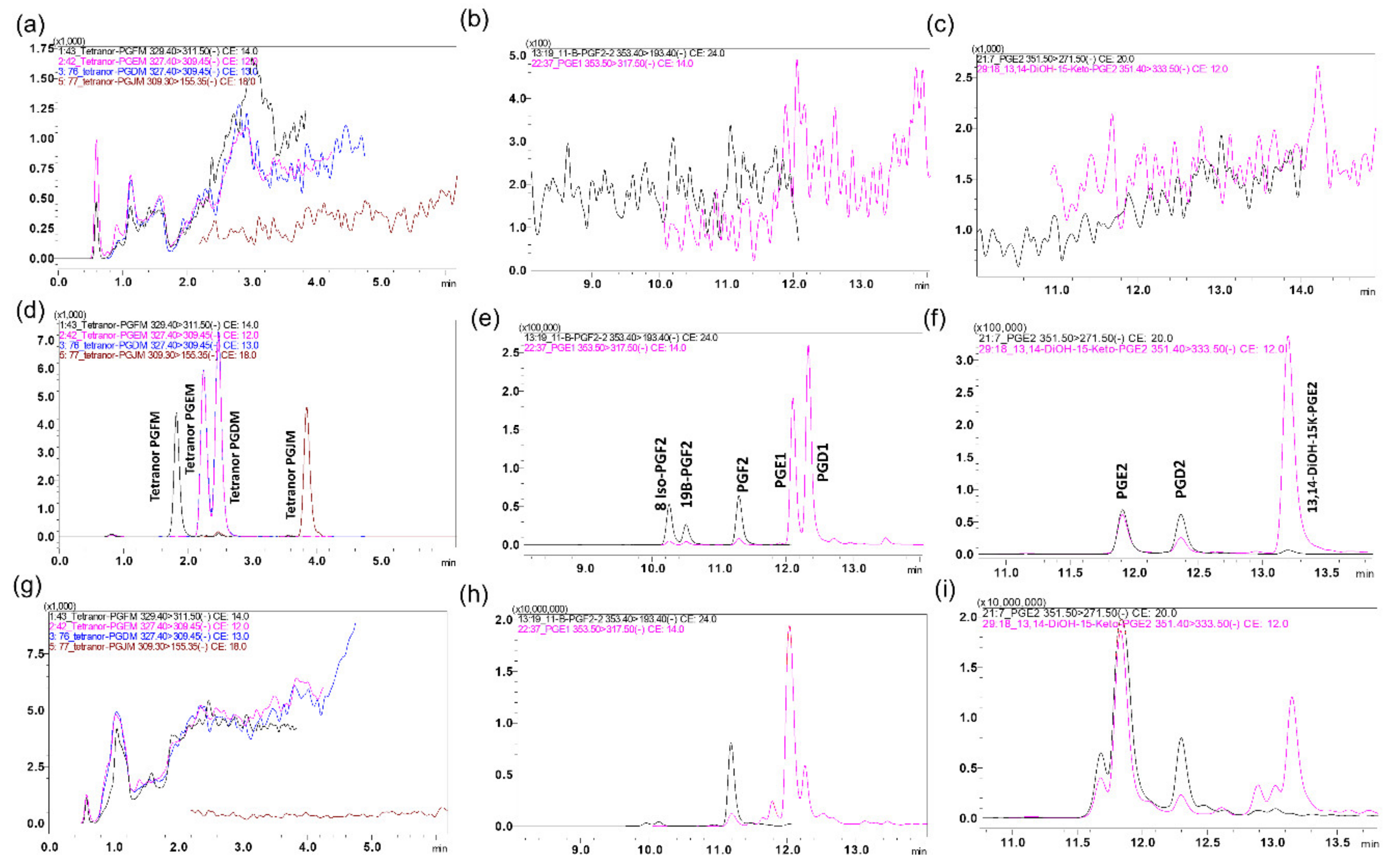
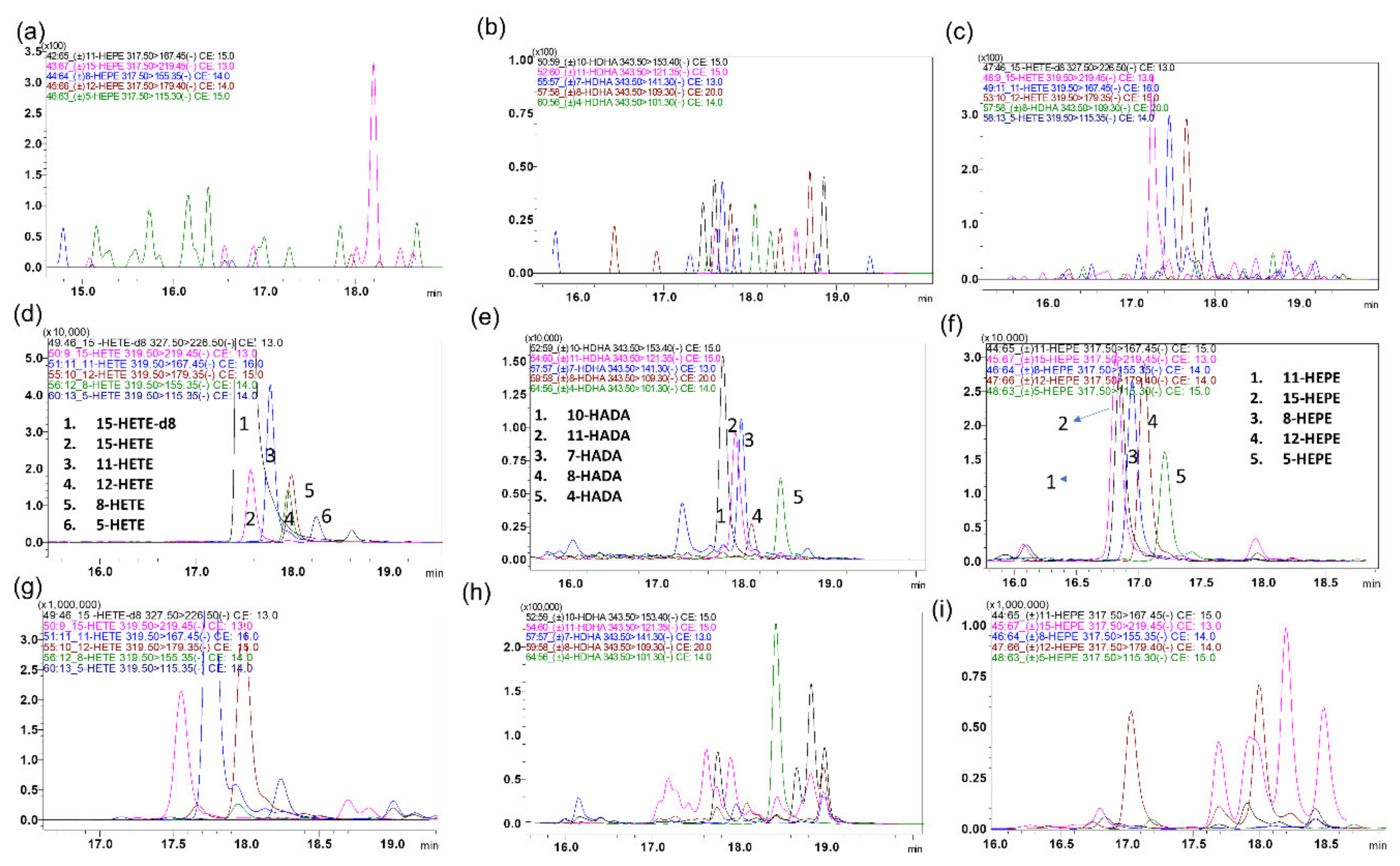
2.1. LC-MS/MS Method Development
2.2. Method Validation
2.3. Recovery and Matrix Effect
2.4. Stability Studies
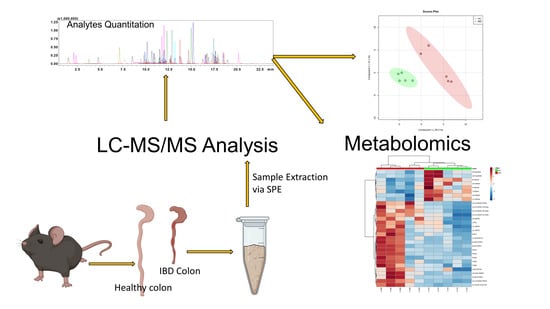
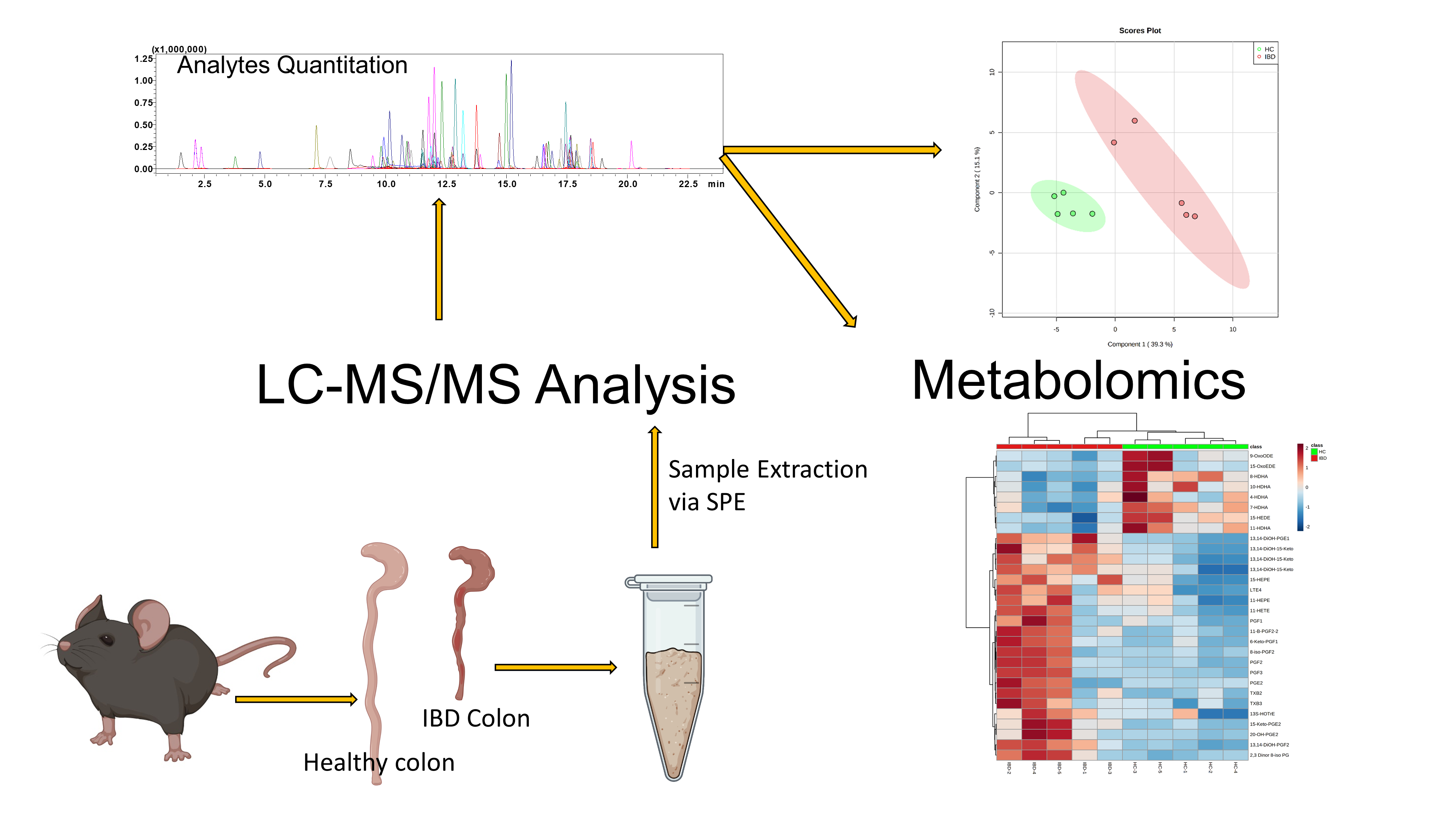
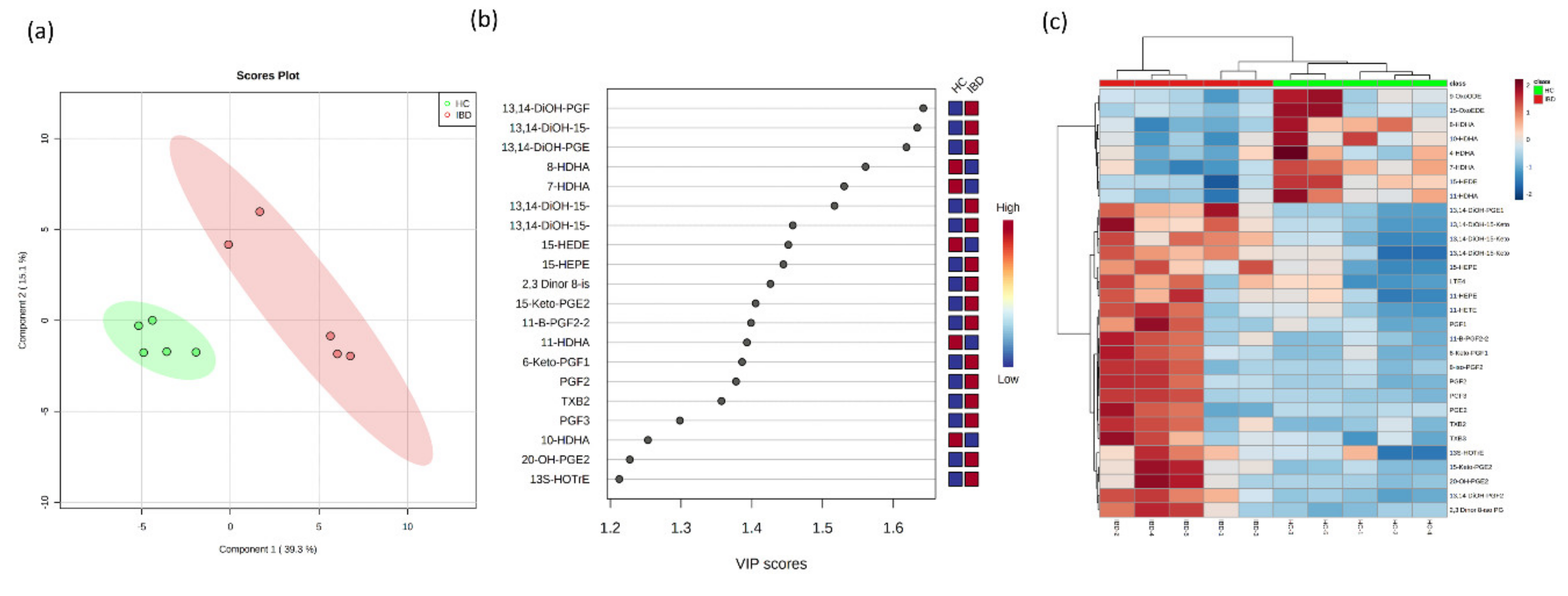
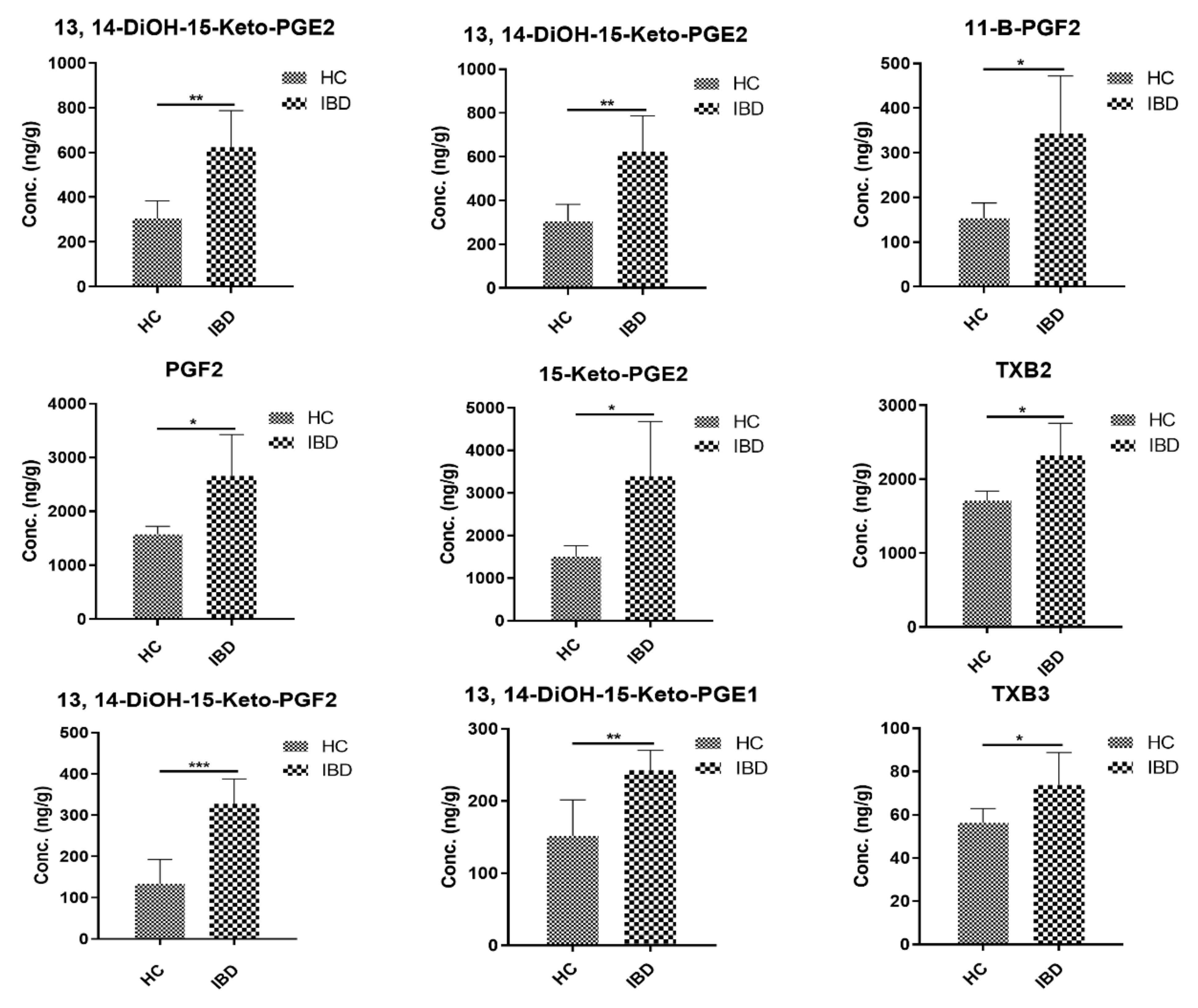
2.5. Application to Metabolomics of Data Profiling
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Liquid Chromatographic and Mass Spectrometric Conditions
3.3. Preparation of Stock, Calibration Standard, and Quality Control Sample Preparation
3.4. Sample Preparation
3.5. Method Validation
3.6. Extraction Recovery and Matrix Effect
3.7. Stability Studies
3.8. Animal Handling, Disease Induction, and Tissue Collection
3.9. Metabolomics Data Processing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. Thematic Review Series: Proteomics. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef] [Green Version]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002, 82, 131–185. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; DuBois, R.N. Measurement of Eicosanoids in Cancer Tissues. Methods Enzymol. 2007, 433, 27–50. [Google Scholar]
- Jain, S.; Chakraborty, G.; Raja, R.; Kale, S.; Kundu, G.C. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Res. 2008, 68, 7750–7759. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, A.H.; Lonigro, A.J.; Hyers, T.M.; Webster, R.O.; Fowler, A.A. Increased concentrations of leukotrienes in bronchoalveolar lavage fluid of patients with ARDS or at risk for ARDS. Am. Rev. Respir. Dis. 1988, 138, 714–719. [Google Scholar] [CrossRef]
- Weiss, J.W.; Drazen, J.M.; McFadden, E.R., Jr.; Weller, P.; Corey, E.J.; Lewis, R.A.; Austen, K.F. Airway constriction in normal humans produced by inhalation of leukotriene D. Potency, time course, and effect of aspirin therapy. J. Am. Med. Assoc. 1983, 249, 2814–2817. [Google Scholar] [CrossRef]
- Miyata, N.; Roman, R.J. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J. Smooth Muscle Res. 2005, 41, 175–193. [Google Scholar] [CrossRef] [Green Version]
- Cambj-Sapunar, L.; Yu, M.; Harder, D.R.; Roman, R.J. Contribution of 5-hydroxytryptamine1B receptors and 20-hydroxyeiscosatetraenoic acid to fall in cerebral blood flow after subarachnoid hemorrhage. Stroke 2003, 34, 1269–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebremedhin, D.; Lange, A.R.; Lowry, T.F.; Taheri, M.R.; Birks, E.K.; Hudetz, A.G.; Narayanan, J.; Falck, J.R.; Okamoto, H.; Roman, R.J.; et al. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ. Res. 2000, 87, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Amaral, S.L.; Maier, K.G.; Schippers, D.N.; Roman, R.J.; Greene, A.S. CYP4A metabolites of arachidonic acid and VEGF are mediators of skeletal muscle angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1528–H1535. [Google Scholar] [CrossRef]
- Amruthesh, S.C.; Falck, J.R.; Ellis, E.F. Brain synthesis and cerebrovascular action of epoxygenase metabolites of arachidonic acid. J. Neurochem. 1992, 58, 503–510. [Google Scholar] [CrossRef]
- Harder, D.R.; Alkayed, N.J.; Lange, A.R.; Gebremedhin, D.; Roman, R.J. Functional hyperemia in the brain: Hypothesis for astrocyte-derived vasodilator metabolites. Stroke 1998, 29, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Fang, X.; Snyder, G.D.; Weintraub, N.L. Epoxyeicosatrienoic acids (EETs): Metabolism and biochemical function. Prog. Lipid Res. 2004, 43, 55–90. [Google Scholar] [CrossRef]
- Iliff, J.J.; Close, L.N.; Selden, N.R.; Alkayed, N.J. A novel role for P450 eicosanoids in the neurogenic control of cerebral blood flow in the rat. Exp. Physiol. 2007, 92, 653–658. [Google Scholar] [PubMed]
- Liu, M.; Alkayed, N.J. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1α-linked induction of P450 2C11 epoxygenase in astrocytes. J. Cereb. Blood Flow Metab. 2005, 25, 939–948. [Google Scholar] [CrossRef]
- Dong, L.M.; Shu, X.O.; Gao, Y.T.; Milne, G.; Ji, B.T.; Yang, G.; Li, H.L.; Rothman, N.; Zheng, W.; Chow, W.H.; et al. Urinary prostaglandin E2 metabolite and gastric cancer risk in the Shanghai women’s health study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3075–3078. [Google Scholar] [CrossRef] [Green Version]
- Eikelboom, J.W.; Hirsh, J.; Weitz, J.I.; Johnston, M.; Yi, Q.; Yusuf, S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002, 105, 1650–1655. [Google Scholar] [CrossRef] [Green Version]
- Gainer, J.V.; Bellamine, A.; Dawson, E.P.; Womble, K.E.; Grant, S.W.; Wang, Y.; Cupples, L.A.; Guo, C.Y.; Demissie, S.; O’Donnell, C.J.; et al. Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 2005, 111, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.C.; Schmidt, C.R.; Shrubsole, M.J.; Billheimer, D.D.; Joshi, P.R.; Morrow, J.D.; Heslin, M.J.; Washington, M.K.; Ness, R.M.; Zheng, W.; et al. Urine PGE-M: A Metabolite of Prostaglandin E2 as a Potential Biomarker of Advanced Colorectal Neoplasia. Clin. Gastroenterol. Hepatol. 2006, 4, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.L.H.; Zhang, Y.; Whitworth, J.A. Reactive oxygen species and glucocorticoid-induced hypertension. Clin. Exp. Pharmacol. Physiol. 2008, 35, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Carrier, D.J.; Bogri, T.; Cosentino, G.P.; Guse, I.; Rakhit, S.; Singh, K. HPLC studies on leukotriene A4 obtained from the hydrolysis of its methyl ester. ProstaglandinsLeukot. Essent. Fat. Acids 1988, 34, 27–30. [Google Scholar] [CrossRef]
- Huwyler, J.; Gut, J. Single-step organic extraction of leukotrienes and related compounds and their simultaneous analysis by high-performance liquid chromatography. Anal. Biochem. 1990, 188, 374–382. [Google Scholar] [CrossRef]
- Lee, K.C.; DeLuca, P.P. Simultanoues determination of prostaglandins E1, A1 and B1 by reversed-phase high-performance liquid chromatography for the kinetic studies of prostaglandin E1 in solution. J. Chromatogr. A 1991, 555, 73–80. [Google Scholar] [CrossRef]
- Shono, F.; Yokota, K.; Horie, K.; Yamamoto, S.; Yamashita, K.; Watanabe, K.; Miyazaki, H. A heterologous enzyme immunoassay of prostaglandin E2 using a stable enzyme-labeled hapten mimic. Anal. Biochem. 1988, 168, 284–291. [Google Scholar] [CrossRef]
- Salmon, J.A. Measurement of eicosanoids by bioassay and radioimmunoassay. Br. Med. Bull. 1983, 39, 227–231. [Google Scholar] [CrossRef] [PubMed]
- VanderNoot, V.A.; VanRollins, M. Capillary Electrophoresis of Cytochrome P-450 Epoxygenase Metabolites of Arachidonic Acid. 1. Resolution of Regioisomers. Anal. Chem. 2002, 74, 5859–5865. [Google Scholar] [CrossRef]
- Herrmann, T.; Steinhilber, D.; Roth, H.J. Determination of leukotriene B4 by high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B Biomed. Sci. Appl. 1987, 416, 170–175. [Google Scholar] [CrossRef]
- Tsikas, D.; Suchy, M.-T.; Tödter, K.; Heeren, J.; Scheja, L. Utilizing immunoaffinity chromatography (IAC) cross-reactivity in GC–MS/MS exemplified at the measurement of prostaglandin E1 in human plasma using prostaglandin E2-specific IAC columns. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1021, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Nithipatikom, K.; Grall, A.J.; Holmes, B.B.; Harder, D.R.; Falck, J.R.; Campbell, W.B. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome p450 metabolites of arachidonic acid. Anal. Biochem. 2001, 298, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Ward, N.; Hodgson, J.; Puddey, I.B.; Falck, J.R.; Croft, K.D. Measurement of 20-Hydroxyeicosatetraenoic Acid in Human Urine by Gas Chromatography-Mass Spectrometry. Clin. Chem. 2004, 50, 224–226. [Google Scholar] [CrossRef]
- Teppner, M.; Zell, M.; Husser, C.; Ernst, B.; Pähler, A. Quantitative profiling of prostaglandins as oxidative stress biomarkers in vitro and in vivo by negative ion online solid phase extraction—Liquid chromatography–tandem mass spectrometry. Anal. Biochem. 2016, 498, 68–77. [Google Scholar] [CrossRef]
- Lubin, A.; Geerinckx, S.; Bajic, S.; Cabooter, D.; Augustijns, P.; Cuyckens, F.; Vreeken, R.J. Enhanced performance for the analysis of prostaglandins and thromboxanes by liquid chromatography-tandem mass spectrometry using a new atmospheric pressure ionization source. J. Chromatogr. A 2016, 1440, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, N.; Ai, D.; Zhu, Y. Systematic Metabolomic Analysis of Eicosanoids after Omega-3 Polyunsaturated Fatty Acid Supplementation by a Highly Specific Liquid Chromatography—Tandem Mass Spectrometry-Based Method. J. Proteome Res. 2015, 14, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, A.I.; Willenberg, I.; Weylandt, K.H.; Schebb, N.H. Development of an Online-SPE–LC–MS/MS Method for 26 Hydroxylated Polyunsaturated Fatty Acids as Rapid Targeted Metabolomics Approach for the LOX, CYP, and Autoxidation Pathways of the Arachidonic Acid Cascade. Chromatographia 2015, 78, 415–428. [Google Scholar] [CrossRef]
- Gachet, M.S.; Rhyn, P.; Bosch, O.G.; Quednow, B.B.; Gertsch, J. A quantitiative LC-MS/MS method for the measurement of arachidonic acid, prostanoids, endocannabinoids, N-acylethanolamines and steroids in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 976–977, 6–18. [Google Scholar] [CrossRef]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; Nelson, A.; Casaburi, R.; Criner, G.; Dransfield, M.T.; Make, B.; Schmid, K.K.; Rennard, S.I.; et al. Simultaneous LC–MS/MS analysis of eicosanoids and related metabolites in human serum, sputum and BALF. Biomed. Chromatogr. 2018, 32, e4102. [Google Scholar] [CrossRef] [PubMed]
- Dumlao, D.S.; Buczynski, M.W.; Norris, P.C.; Harkewicz, R.; Dennis, E.A. High-throughput lipidomic analysis of fatty acid derived eicosanoids and N-acylethanolamines. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 724–736. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Liu, X.; Wu, J.; Meehan, M.J.; Blevitt, J.M.; Dorrestein, P.C.; Milla, M.E. A highly efficient, high-throughput lipidomics platform for the quantitative detection of eicosanoids in human whole blood. Anal. Biochem. 2013, 433, 181–188. [Google Scholar] [CrossRef]
- Wang, Y.; Armando, A.M.; Quehenberger, O.; Yan, C.; Dennis, E.A. Comprehensive ultra-performance liquid chromatographic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. J. Chromatogr. A 2014, 1359, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Hu, T.; An, Z.; Li, P.; Liu, L. Simultaneous quantitative determination of arachidonic acid and cascade metabolites in rat serum by UPLC-MS/MS: Application for longitudinal metabolomics of anlotinib. Analyst 2020, 145, 4972–4981. [Google Scholar] [CrossRef]
- Silva, F.A.R.; Rodrigues, B.L.; de Ayrizono, M.L.S.; Leal, R.F. The Immunological Basis of Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2016, 2016, 2097274. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.J. Eicosanoid receptors: Targets for the treatment of disrupted intestinal epithelial homeostasis. Eur. J. Pharmacol. 2017, 796, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, M.; Nicolaou, A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom 2006, 20, 3023–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempen, E.C.; Yang, P.; Felix, E.; Madden, T.; Newman, R.A. Simultaneous Quantification of Arachidonic Acid Metabolites in Cultured Tumor Cells Using High-Performance Liquid Chromatography/Electrospray Ionization Tandem Mass Spectrometry. Anal. Biochem. 2001, 297, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Gomolka, B.; Siegert, E.; Blossey, K.; Schunck, W.H.; Rothe, M.; Weylandt, K.H. Analysis of omega-3 and omega-6 fatty acid-derived lipid metabolite formation in human and mouse blood samples. Prostaglandins Other Lipid Mediat. 2011, 94, 81–87. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Tackett, R.L.; Stewart, J.T.; Bartlett, M.G. Determination of arginine and methylated arginines in human plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 2000, 748, 157–166. [Google Scholar] [CrossRef]
- Yang, J.; Schmelzer, K.; Georgi, K.; Hammock, B.D. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Chem. 2009, 81, 8085–8093. [Google Scholar] [CrossRef] [Green Version]
- Prasain, J.K.; Arabshahi, A.; Taub, P.R.; Sweeney, S.; Moore, R.; Sharer, J.D.; Barnes, S. Simultaneous quantification of F2-isoprostanes and prostaglandins in human urine by liquid chromatography tandem-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 913–914, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Tomaru, K.; Matsumoto, N.; Watanabe, S.; Higashi, T. LC/ESI-MS/MS method for determination of salivary eicosapentaenoic acid concentration to arachidonic acid concentration ratio. Biomed. Chromatogr. 2015, 30, 29–34. [Google Scholar] [CrossRef]
- Deems, R.; Buczynski, M.W.; Bowers-Gentry, R.; Harkewicz, R.; Dennis, E.A. Detection and Quantitation of Eicosanoids via High Performance Liquid Chromatography-Electrospray Ionization-Mass Spectrometry. In Methods in Enzymology; Brown, H.A., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 432, pp. 59–82. [Google Scholar]
- Levison, B.S.; Zhang, R.; Wang, Z.; Fu, X.; Didonato, J.A.; Hazen, S.L. Quantification of fatty acid oxidation products using online high-performance liquid chromatography tandem mass spectrometry. Free Radic. Biol. Med. 2013, 59, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouveia-Figueira, S.; Nording, M.L. Validation of a tandem mass spectrometry method using combined extraction of 42 oxylipins and 15 endocannabinoid-related compounds including prostamides from biological matrices. Prostaglandins Other Lipid Mediat. 2015, 121, 110–121. [Google Scholar] [CrossRef]
- Montuschi, P.; Martello, S.; Felli, M.; Mondino, C.; Chiarotti, M. Ion trap liquid chromatography/tandem mass spectrometry analysis of leukotriene B4 in exhaled breath condensate. Rapid Commun. Mass Spectrom. 2004, 18, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Idborg, H.; Pawelzik, S.-C.; Perez-Manso, M.; Björk, L.; Hamrin, J.; Herlenius, E.; Jakobsson, P.-J. Evaluation of urinary prostaglandin E2 metabolite as a biomarker in infants with fever due to viral infection. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Squellerio, I.; Porro, B.; Songia, P.; Veglia, F.; Caruso, D.; Tremoli, E.; Cavalca, V. Liquid chromatography-tandem mass spectrometry for simultaneous measurement of thromboxane B2 and 12(S)-hydroxyeicosatetraenoic acid in serum. J. Pharm. Biomed. Anal. 2014, 96, 256–262. [Google Scholar] [CrossRef]
- Massey, K.A.; Nicolaou, A. Lipidomics of oxidized polyunsaturated fatty acids. Free Radic. Biol. Med. 2013, 59, 45–55. [Google Scholar] [CrossRef]
- Kortz, L.; Dorow, J.; Becker, S.; Thiery, J.; Ceglarek, U. Fast liquid chromatography-quadrupole linear ion trap-mass spectrometry analysis of polyunsaturated fatty acids and eicosanoids in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 927, 209–213. [Google Scholar] [CrossRef]
- Jian, W.; Edom, R.W.; Xue, X.; Huang, M.Q.; Fourie, A.; Weng, N. Quantitation of leukotriene B4 in human sputum as a biomarker using UPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 932, 59–65. [Google Scholar] [CrossRef]
- Yoshida, Y.; Kodai, S.; Takemura, S.; Minamiyama, Y.; Niki, E. Simultaneous measurement of F2-isoprostane, hydroxyoctadecadienoic acid, hydroxyeicosatetraenoic acid, and hydroxycholesterols from physiological samples. Anal. Biochem. 2008, 379, 105–115. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, G.; Clarke, P.A.; Huang, J.T.J.; Takahashi, E.; Muirhead, D.; Steenwyk, R.C.; Lin, Z. Simultaneous and high-throughput quantitation of urinary tetranor PGDM and tetranor PGEM by online SPE-LC–MS/MS as inflammatory biomarkers. J. Mass Spectrom. 2011, 46, 705–711. [Google Scholar] [CrossRef]
- Donowitz, M. Arachidonic acid metabolites and their role in inflammatory bowel disease. Gastroenterology 1985, 88, 580–587. [Google Scholar] [CrossRef]
- Higgins, A.J.; Lees, P. The acute inflammatory process, arachidonic acid metabolism and the mode of action of anti-inflammatory drugs. Equine Vet. J. 2010, 16, 163–175. [Google Scholar] [CrossRef]
- Malmsten, C.L. Prostaglandins, thromboxanes, and leukotrienes in inflammation. Semin. Arthritis Rheum. 1985, 15, 29–35. [Google Scholar] [CrossRef]
- Gould, S.R. Assay of prostaglandin-like substances in faeces and their measurement in ulcerative colitis. Prostaglandins 1976, 11, 489–497. [Google Scholar] [CrossRef]
- Ligumsky, M.; Karmeli, F.; Sharon, P.; Zor, U.; Cohen, F.; Rachmilewitz, D. Enhanced thromboxane A2 and prostacyclin production by cultured rectal mucosa in ulcerative colitis and its inhibition by steroids and sulfasalazine. Gastroenterology 1981, 81, 444–449. [Google Scholar] [CrossRef]
- Kuehl, F.A.; Egan, R.W. Prostaglandins, arachidonic acid, and inflammation. Science 1980, 210, 978. [Google Scholar] [CrossRef]
- Chen, I.-J.; Hee, S.-W.; Liao, C.-H.; Lin, S.-Y.; Su, L.; Shun, C.-T.; Chuang, L.-M. Targeting the 15-keto-PGE2-PTGR2 axis modulates systemic inflammation and survival in experimental sepsis. Free Radic. Biol. Med. 2018, 115, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Matsumoto, M.; Takeda, K.; Akira, S. Lipopolysaccharide-Dependent Prostaglandin E(2) Production Is Regulated by the Glutathione-Dependent Prostaglandin E(2) Synthase Gene Induced by the Toll-Like Receptor 4/MyD88/NF-IL6 Pathway. J. Immunol. 2002, 168, 5811. [Google Scholar] [CrossRef] [Green Version]
- Sheibanie, A.F.; Yen, J.-H.; Khayrullina, T.; Emig, F.; Zhang, M.; Tuma, R.; Ganea, D. The Proinflammatory Effect of Prostaglandin E(2) in Experimental Inflammatory Bowel Disease Is Mediated through the IL-23→IL-17 Axis. J. Immunol. 2007, 178, 8138. [Google Scholar] [CrossRef] [Green Version]
- Kirkby Shaw, K.; Rausch-Derra, L.C.; Rhodes, L. Grapiprant: An EP4 prostaglandin receptor antagonist and novel therapy for pain and inflammation. Vet. Med. Sci. 2016, 2, 3–9. [Google Scholar] [CrossRef]
- Lowe, N.J.; Virgadamo, F.; Stoughton, R.B. Anti-inflammatory properties of a prostaglandin antagonist, a corticosteroid and indomethacin in experimental contact dermatitis. Br. J. Dermatol. 1977, 96, 433–438. [Google Scholar] [CrossRef]
- Basu, S.; Whiteman, M.; Mattey, D.L.; Halliwell, B. Raised levels of F2-isoprostanes and prostaglandin F2α in different rheumatic diseases. Ann. Rheum. Dis. 2001, 60, 627. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, H.; Sugiyama, S.; Ohara, A.; Goto, H.; Tsukamoto, Y.; Ozawa, T. Mechanism and prevention of chronic colonic inflammation with trinitrobenzene sulfonic acid in rats. Clin. Exp. Pharmacol. Physiol. 2007, 19, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Basu, S. Oxidative Injury Induced Cyclooxygenase Activation in Experimental Hepatotoxicity. Biochem. Biophys. Res. Commun. 1999, 254, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, G.; Nizankowska, E.; Gielicz, A.; Swierczynska, M.; Szczeklik, A. Plasma 9α,11ß-PGF2, a PGD2 metabolite, as a sensitive marker of mast cell activation by allergen in bronchial asthma. Thorax 2004, 59, 459–464. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Aritake, K.; Tsubosaka, Y.; Maruyama, T.; Nakagawa, T.; Hori, M.; Hirai, H.; Nakamura, M.; Narumiya, S.; Urade, Y.; et al. Anti-inflammatory role of PGD2 in acute lung inflammation and therapeutic application of its signal enhancement. Proc. Natl. Acad. Sci. USA 2013, 110, 5205. [Google Scholar] [CrossRef] [Green Version]
- Rampton, D.S.; Collins, C.E. Thrornboxanes in inflammatory bowel disease—pathogenic and therapeutic implications. Aliment. Pharmacol. Ther. 1993, 7, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Carty, E.; Nickols, C.; Feakins, R.M.; Rampton, D.S. Thromboxane synthase immunohistochemistry in inflammatory bowel disease. J. Clin. Pathol. 2002, 55, 367–370. [Google Scholar] [CrossRef]
- Tozaki, H.; Fujita, T.; Odoriba, T.; Terabe, A.; Suzuki, T.; Tanaka, C.; Okabe, S.; Muranishi, S.; Yamamoto, A. Colon-specific delivery of R68070, a new thromboxane synthase inhibitor, using chitosan capsules: Therapeutic effects against 2,4,6-trinitrobenzene sulfonic acid-induced ulcerative colitis in rats. Life Sci. 1999, 64, 1155–1162. [Google Scholar] [CrossRef]
- Gould, S.R.; Brash, A.R.; Conolly, M.E.; Lennard-Jones, J.E. Studies of prostaglandins and sulphasalazine in ulcerative colitis. Prostaglandins Med. 1981, 6, 165–182. [Google Scholar] [CrossRef]
- Paruchuri, S.; Tashimo, H.; Feng, C.; Maekawa, A.; Xing, W.; Jiang, Y.; Kanaoka, Y.; Conley, P.; Boyce, J.A. Leukotriene E4–induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 2009, 206, 2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauler, J.; Thon, A.; Tsikas, D.; Hardt, H.V.D.; Frölich, J.C. Enhanced Synthesis of Cysteinyl Leukotrienes in Juvenile Rheumatoid Arthritis. Arthritis Rheum. 1994, 37, 93–97. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Pofelski, J.; Moreau-Gaudry, A.; Bessard, G.; Bonaz, B. Urinary leukotriene E4 excretion: A biomarker of inflammatory bowel disease activity. Inflamm. Bowel Dis. 2008, 14, 769–774. [Google Scholar] [CrossRef]
- Jupp, J.; Hillier, K.; Elliott, D.H.; Fine, D.R.; Bateman, A.C.; Johnson, P.A.; Cazaly, A.M.; Penrose, J.F.; Sampson, A.P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 537–546. [Google Scholar] [CrossRef]
- Chhonker, Y.S.; Haney, S.L.; Bala, V.; Holstein, S.A.; Murry, D.J. Simultaneous Quantitation of Isoprenoid Pyrophosphates in Plasma and Cancer Cells Using LC-MS/MS. Molecules 2018, 23, 3275. [Google Scholar] [CrossRef] [Green Version]
- FDA, Drug Administration Centre for Drug Evaluation and Research (FDA). Guidance for Industry-Bioanalytical Method Validation; US Department for Health and Human Services—Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2018.
- Bhinder, G.; Sham, H.P.; Chan, J.M.; Morampudi, V.; Jacobson, K.; Vallance, B.A. The Citrobacter rodentium Mouse Model: Studying Pathogen and Host Contributions to Infectious Colitis. J. Vis. Exp. JoVE 2013, 72, e50222. [Google Scholar] [CrossRef] [Green Version]
- Kanvinde, S.; Chhonker, Y.S.; Ahmad, R.; Yu, F.; Sleightholm, R.; Tang, W.; Jaramillo, L.; Chen, Y.; Sheinin, Y.; Li, J.; et al. Pharmacokinetics and efficacy of orally administered polymeric chloroquine as macromolecular drug in the treatment of inflammatory bowel disease. Acta Biomater. 2018, 82, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes ID | Analytes | MRM (m/z) | RT (Min.) | Q1 Pre Vias (V) | CE (V) | Q3 Pre Vias (V) | Dynamic Range (ng/mL) |
|---|---|---|---|---|---|---|---|
| 1 | PGJ2 | 333.50 > 233.40 | 13.7 | 12 | 10 | 15 | 0.01–200 |
| 3 | 20-OH-PGE2 | 367.40 > 287.45 | 4.9 | 12 | 17 | 19 | 0.01–200 |
| 4 | PGB2 | 333.50 > 235.45 | 13.9 | 11 | 19 | 15 | 0.01–200 |
| 6 | PGD2 | 351.50 > 271.50 | 12.0 | 12 | 17 | 17 | 0.01–200 |
| 7 | PGE2 | 351.50 > 271.50 | 11.6 | 12 | 17 | 17 | 0.01–200 |
| 8 | AA | 303.50 > 259.50 | 20.6 | 22 | 14 | 11 | 1–200 |
| 9 | 15-HETE | 319.50 > 219.45 | 17.3 | 11 | 14 | 13 | 0.01–200 |
| 10 | 12-HETE | 319.50 > 179.35 | 17.7 | 11 | 15 | 10 | 0.01–200 |
| 11 | 11-HETE | 319.50 > 167.45 | 17.5 | 11 | 16 | 10 | 0.01–200 |
| 12 | 8-HETE | 319.50 > 155.35 | 17.7 | 16 | 14 | 24 | 0.01–200 |
| 13 | 5-HETE | 319.50 > 115.35 | 18.0 | 11 | 14 | 16 | 0.01–200 |
| 14 | LTE4 | 438.40 > 235.45 | 12.8 | 10 | 23 | 14 | 1–200 |
| 15 | LTD4 | 495.50 > 177.35 | 11.0 | 11 | 20 | 10 | 1–200 |
| 16 | LTC4 | 624.50 > 272.40 | 12.0 | 22 | 24 | 12 | 1–200 |
| 17 | LTB4 | 335.50 > 195.40 | 14.7 | 12 | 16 | 11 | 0.01–200 |
| 18 | 13,14-DiOH-15-Keto-PGE2 | 351.40 > 333.50 | 12.9 | 11 | 12 | 10 | 0.01–200 |
| 19 | 11-B-PGF2 | 353.40 > 193.40 | 10.1 | 12 | 24 | 12 | 0.01–200 |
| 20 | 8-iso-PGF2 | 353.40 > 193.40 | 9.8 | 12 | 24 | 12 | 0.01–200 |
| 21 | PGF2 | 353.40 > 193.40 | 10.9 | 12 | 24 | 12 | 0.01–200 |
| 22 | 15-Keto-PGE2 | 349.30 > 235.40 | 12.3 | 18 | 15 | 15 | 0.01–200 |
| 23 | 6-Keto-PGF1 | 369.40 > 245.45 | 7.7 | 12 | 25 | 15 | 0.01–200 |
| 24 | TXB2 | 369.50 > 169.40 | 10.0 | 13 | 18 | 10 | 0.01–200 |
| 29 | 13,14-DiOH-PGF2 | 355.40 > 311.50 | 12.0 | 11 | 25 | 14 | 0.01–200 |
| 30 | PGF1 | 355.40 > 311.50 | 11.0 | 11 | 25 | 14 | 0.01–200 |
| 31 | 13,14-DiOH-15-Keto-PGF2 | 353.50 > 113.40 | 12.7 | 12 | 25 | 11 | 0.01–200 |
| 32 | 13,14-DiOH-15-Keto-PGE1 | 353.40 > 335.50 | 13.2 | 12 | 13 | 22 | 0.01–200 |
| 33 | PGD1 | 353.50 > 317.50 | 12.1 | 12 | 17 | 15 | 0.01–200 |
| 34 | 13,14-DiOH-PGE1 | 355.30 > 337.55 | 12.4 | 11 | 16 | 11 | 0.01–200 |
| 35 | TXB3 | 367.50 > 169.35 | 8.6 | 10 | 17 | 10 | 0.01–200 |
| 36 | 15-deoxy-delta 12,14 PGJ2 | 315.50 > 271.50 | 13.8 | 12 | 14 | 17 | 0.01–200 |
| 37 | PGE1 | 353.50 > 317.50 | 11.8 | 12 | 17 | 15 | 0.01–200 |
| 38 | PGE3 | 349.50 > 269.50 | 10.2 | 12 | 16 | 17 | 0.01–200 |
| 39 | PGD3 | 349.50 > 269.50 | 10.7 | 12 | 16 | 17 | 0.01–200 |
| 40 | PGF3 | 351.50 > 307.50 | 9.5 | 12 | 18 | 14 | 0.1–200 |
| 41 | 14,15-LTC4 | 624.50 > 272.40 | 13.0 | 22 | 23 | 18 | 1–200 |
| 42 | Tetranor-PGEM | 327.40 > 309.45 | 2.2 | 11 | 12 | 21 | 0.1–200 |
| 43 | Tetranor-PGFM | 329.40 > 311.50 | 1.8 | 16 | 14 | 10 | 0.1–200 |
| 44 | 11-De TXB3 | 365.40 > 303.50 | 10.4 | 13 | 16 | 20 | 0.01–200 |
| 45 | 2,3 Dinor 8-iso PGF2 | 325.50 > 237.50 | 7.2 | 12 | 13 | 15 | 0.01–200 |
| 53 | Docosahexaenoic Acid | 327.50 > 283.55 | 20.3 | 11 | 12 | 18 | 1–200 |
| 54 | 9(10)-DiHOME | 313.50 > 201.40 | 15.2 | 11 | 22 | 12 | 0.01–200 |
| 55 | 12(13)-DiHOME | 313.50 > 183.40 | 15.1 | 11 | 22 | 11 | 0.01–200 |
| 56 | 4-HDHA | 343.50 > 101.30 | 18.1 | 11 | 14 | 29 | 0.01–200 |
| 57 | 7-HDHA | 343.50 > 141.30 | 17.7 | 12 | 13 | 23 | 0.01–200 |
| 58 | 8-HDHA | 343.50 > 109.30 | 17.8 | 11 | 20 | 30 | 0.01–200 |
| 59 | 10-HDHA | 343.50 > 153.40 | 17.5 | 12 | 15 | 25 | 0.01–200 |
| 60 | 11-HDHA | 343.50 > 121.35 | 17.7 | 12 | 15 | 18 | 0.01–200 |
| 61 | 11-HEDE | 323.40 > 199.45 | 18.5 | 11 | 21 | 12 | 0.01–200 |
| 62 | 15-HEDE | 323.40 > 223.50 | 18.6 | 11 | 20 | 14 | 0.01–200 |
| 63 | 5-HEPE | 317.50 > 115.30 | 17.0 | 12 | 15 | 18 | 0.01–200 |
| 64 | 8-HEPE | 317.50 > 155.35 | 16.7 | 10 | 14 | 26 | 0.01–200 |
| 65 | 11-HEPE | 317.50 > 167.45 | 16.6 | 11 | 15 | 10 | 0.01–200 |
| 66 | 12-HEPE | 317.50 > 179.40 | 16.8 | 12 | 14 | 10 | 0.01–200 |
| 67 | 15-HEPE | 317.50 > 219.45 | 16.7 | 12 | 13 | 13 | 0.01–200 |
| 68 | 9(S)-HOTrE | 293.50 > 171.35 | 16.2 | 12 | 15 | 10 | 0.01–200 |
| 69 | 13(S)-HOTrE | 293.50 > 224.45 | 16.3 | 11 | 14 | 13 | 0.01–200 |
| 70 | 5-OxoETE | 317.50 > 203.40 | 18.6 | 10 | 18 | 12 | 0.01–200 |
| 71 | 12-OxoETE | 317.50 > 153.40 | 18.0 | 16 | 16 | 25 | 0.01–200 |
| 72 | 15-OxoETE or 15-KETE | 317.50 > 113.35 | 17.7 | 12 | 20 | 18 | 0.01–200 |
| 73 | 9-OxoODE or 9-KEDE | 293.50 > 185.40 | 17.8 | 11 | 21 | 11 | 0.01–200 |
| 74 | 15-OxoEDE or 15-KEDE | 321.50 > 223.45 | 19.1 | 11 | 23 | 14 | 0.01–200 |
| 76 | Tetranor-PGDM | 327.40 > 309.45 | 2.8 | 11 | 13 | 20 | 0.1–200 |
| 77 | Tetranor-PGJM | 309.30 > 155.35 | 4.2 | 12 | 22 | 24 | 0.01–200 |
| 78 | Resolvin D1 | 375.60 > 141.30 | 12.9 | 10 | 15 | 23 | 1–200 |
| 79 | Resolvin D2 | 375.60 > 175.35 | 12.2 | 10 | 23 | 10 | 0.1–200 |
| 80 | Resolvin D3 | 375.60 > 147.40 | 12.0 | 10 | 22 | 23 | 0.1–200 |
| 25 | TXB2-d4 | 373.50 > 173.40 | 10.0 | 13 | 19 | 10 | NA |
| 26 | PGE2-d4 | 355.50 > 275.55 | 11.5 | 13 | 17 | 18 | NA |
| 27 | AA-d8 | 311.40 > 267.55 | 20.6 | 10 | 13 | 17 | NA |
| 46 | 15 -HETE-d8 | 327.50 > 226.50 | 17.2 | 16 | 13 | 14 | NA |
| 47 | LTB4-d4 | 339.50 > 197.45 | 14.7 | 10 | 16 | 12 | NA |
| 81 | Resolvin D1-d5 | 380.50 > 141.30 | 12.8 | 13 | 16 | 22 | NA |
| Analytes ID | LLOQ (0.01 ng/mL) | LQC (0.05 ng/mL) | MQC (40 ng/mL) | HQC (150 ng/mL) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Accuracy | %RSD | Accuracy | %RSD | Accuracy | %RSD | Accuracy | %RSD | ||
| 1 | PGJ2 | 108.9 | 14.3 | 106.6 | 2.6 | 103.7 | 4.0 | 86.9 | 3.9 |
| 3 | 20-OH-PGE2 | 112.6 | 5.1 | 92.4 | 5.5 | 104.0 | 6.2 | 108.9 | 0.5 |
| 4 | PGB2 | 114.9 | 4.9 | 99.7 | 9.1 | 101.7 | 4.9 | 86.8 | 1.9 |
| 6 | PGD2 | 103.9 | 11.2 | 108.7 | 3.2 | 92.5 | 2.8 | 90.6 | 4.9 |
| 7 | PGE2 | 99.2 | 10.0 | 98.1 | 1.8 | 97.8 | 5.6 | 87.3 | 3.7 |
| 9 | 15-HETE | 100.7 | 17.3 | 112.7 | 3.8 | 96.7 | 6.0 | 85.3 | 1.3 |
| 10 | 12-HETE | 104.6 | 5.4 | 108.4 | 4.8 | 96.9 | 1.4 | 87.5 | 3.7 |
| 11 | 11-HETE | 104.5 | 5.6 | 114.5 | 3.2 | 87.1 | 1.4 | 91.1 | 3.3 |
| 12 | 8-HETE | 190.5 | 14.8 | 114.7 | 4.3 | 92.1 | 5.5 | 84.7 | 5.8 |
| 13 | 5-HETE | 109.9 | 14.3 | 110.4 | 3.4 | 103.1 | 2.9 | 87.0 | 0.9 |
| 17 | LTB4 | 112.5 | 18.4 | 110.1 | 6.6 | 92.0 | 5.9 | 103.8 | 6.1 |
| 18 | 13,14-DiOH-15-Keto-PGE2 | 102.3 | 11.2 | 107.2 | 3.2 | 97.8 | 2.5 | 90.2 | 2.2 |
| 19 | 11-B-PGF2 | 107.9 | 18.2 | 106.7 | 1.4 | 105.8 | 4.3 | 87.3 | 0.8 |
| 20 | 8-iso-PGF2 | 112.2 | 9.1 | 103.3 | 4.5 | 103.9 | 5.6 | 88.3 | 2.3 |
| 21 | PGF2 | 110.2 | 9.1 | 104.0 | 4.5 | 100.6 | 4.3 | 88.0 | 2.1 |
| 22 | 15-Keto-PGE2 | 101.3 | 11.2 | 107.5 | 2.7 | 101.9 | 2.5 | 91.0 | 2.8 |
| 23 | 6-Keto-PGF1 | 90.9 | 19.2 | 92.0 | 6.1 | 107.6 | 5.1 | 107.7 | 1.6 |
| 24 | TXB2 | 109.6 | 15.7 | 108.0 | 4.6 | 90.1 | 5.1 | 86.5 | 2.5 |
| 29 | 13,14-DiOH-PGF2 | 107.3 | 5.4 | 109.9 | 8.9 | 104.7 | 4.3 | 107.8 | 1.7 |
| 30 | PGF1 | 90.1 | 19.2 | 106.1 | 8.9 | 101.5 | 3.1 | 89.1 | 2.9 |
| 31 | 13,14-DiOH-15-Keto-PGF2 | 116.6 | 10.2 | 105.7 | 5.1 | 101.4 | 4.2 | 88.7 | 3.4 |
| 32 | 13,14-DiOH-15-Keto-PGE1 | 110.8 | 9.1 | 99.9 | 5.5 | 102.5 | 3.8 | 95.7 | 3.7 |
| 33 | PGD1 | 101.2 | 10.0 | 115.1 | 3.2 | 92.2 | 4.1 | 88.1 | 3.2 |
| 34 | 13,14-DiOH-PGE1 | 107.7 | 5.4 | 109.6 | 4.7 | 100.5 | 4.4 | 90.9 | 3.0 |
| 35 | TXB3 | 91.3 | 11.1 | 98.6 | 3.7 | 93.2 | 3.0 | 105.5 | 3.8 |
| 36 | 15-deoxy-delta 12,14 PGJ2 | 98.4 | 15.8 | 85.6 | 16.1 | 100.9 | 2.8 | 88.0 | 3.1 |
| 37 | PGE1 | 86.0 | 18.3 | 104.8 | 3.9 | 100.2 | 3.8 | 87.2 | 3.2 |
| 38 | PGE3 | 99.0 | 6.0 | 105.8 | 6.7 | 99.4 | 2.5 | 85.6 | 1.6 |
| 39 | PGD3 | 90.4 | 3.1 | 99.8 | 2.3 | 97.3 | 0.2 | 93.0 | 2.2 |
| 44 | 11-De TXB3 | 85.6 | 6.9 | 104.2 | 9.8 | 96.7 | 2.6 | 91.3 | 1.1 |
| 45 | 2,3 Dinor 8-iso PGF2 | 107.4 | 9.1 | 95.1 | 2.5 | 102.8 | 6.6 | 102.3 | 2.1 |
| 54 | 9(10)-DiHOME | 109.6 | 9.1 | 113.2 | 2.6 | 90.7 | 4.7 | 86.8 | 1.4 |
| 55 | 12(13)-DiHOME | 112.1 | 18.4 | 112.3 | 2.2 | 100.6 | 2.0 | 91.7 | 5.7 |
| 56 | 4-HDHA | 119.4 | 4.7 | 104.2 | 7.4 | 105.2 | 6.1 | 86.9 | 1.0 |
| 57 | 7-HDHA | 112.9 | 15.7 | 107.3 | 16.5 | 85.1 | 9.6 | 86.0 | 4.8 |
| 58 | 8-HDHA | 118.1 | 16.4 | 104.9 | 7.3 | 103.8 | 5.4 | 94.9 | 3.2 |
| 59 | 10-HDHA | 118.4 | 18.9 | 112.0 | 3.6 | 89.3 | 4.1 | 85.9 | 5.5 |
| 60 | 11-HDHA | 109.3 | 21.7 | 104.5 | 5.9 | 103.7 | 5.6 | 91.1 | 1.6 |
| 61 | 11-HEDE | 107.1 | 14.8 | 109.5 | 9.1 | 87.3 | 3.0 | 84.2 | 8.0 |
| 62 | 15-HEDE | 114.6 | 5.1 | 109.2 | 3.3 | 92.0 | 5.4 | 86.6 | 5.3 |
| 63 | 5-HEPE | 107.2 | 19.5 | 107.5 | 5.7 | 110.8 | 3.0 | 88.6 | 1.2 |
| 64 | 8-HEPE | 100.2 | 20.0 | 114.9 | 4.0 | 101.6 | 11.5 | 91.7 | 2.1 |
| 65 | 11-HEPE | 118.1 | 4.9 | 106.3 | 9.4 | 89.1 | 3.3 | 87.4 | 5.6 |
| 66 | 12-HEPE | 119.1 | 4.7 | 107.9 | 4.0 | 105.7 | 3.4 | 88.0 | 3.2 |
| 67 | 15-HEPE | 109.1 | 15.7 | 111.0 | 4.1 | 92.2 | 5.9 | 85.6 | 2.6 |
| 68 | 9(S)-HOTrE | 110.3 | 18.2 | 109.4 | 7.2 | 111.6 | 7.4 | 85.9 | 1.1 |
| 69 | 13(S)-HOTrE | 105.4 | 19.5 | 111.1 | 1.4 | 108.2 | 4.5 | 86.7 | 3.8 |
| 70 | 5-OxoETE | 99.2 | 5.6 | 98.8 | 4.5 | 105.8 | 8.1 | 90.1 | 1.4 |
| 71 | 12-OxoETE | 104.2 | 20.2 | 107.4 | 9.1 | 88.7 | 4.3 | 86.0 | 18.8 |
| 72 | 15-OxoETE or 15-KETE | 120.0 | 8.3 | 114.7 | 8.7 | 96.5 | 3.7 | 85.3 | 4.7 |
| 73 | 9-OxoODE or 9-KEDE | 122.2 | 4.7 | 112.9 | 2.5 | 104.5 | 5.0 | 89.3 | 4.6 |
| 74 | 15-OxoEDE or 15-KEDE | 103.8 | 20.1 | 104.8 | 3.1 | 100.3 | 3.1 | 89.7 | 3.8 |
| 77 | Tetranor-PGJM | 83.9 | 6.7 | 103.0 | 4.2 | 113.3 | 6.2 | 114.9 | 3.2 |
| 78 | Resolvin D1 | 108.7 | 19.5 | 103.7 | 14.0 | 104.6 | 3.5 | 89.7 | 2.6 |
| LLOQ (0.05 ng/mL) | LQC (2 ng/mL) | MQC (40 ng/mL) | HQC (150 ng/mL) | ||||||
| 40 | PGF3 | 99.2 | 14.1 | 99.6 | 6.6 | 103.7 | 6.6 | 102.4 | 1.7 |
| 42 | Tetranor-PGEM | 120.4 | 1.0 | 111.7 | 14.2 | 89.5 | 12.3 | 96.7 | 2.4 |
| 43 | Tetranor-PGFM | 109.8 | 20.1 | 85.9 | 4.2 | 99.3 | 3.5 | 100.5 | 6.3 |
| 76 | Tetranor-PGDM | 98.5 | 6.3 | 86.4 | 5.0 | 113.8 | 8.0 | 114.7 | 2.7 |
| 79 | Resolvin D2 | 105.5 | 15.4 | 102.6 | 12.3 | 96.0 | 6.7 | 89.1 | 0.3 |
| 80 | Resolvin D3 | 106.7 | 11.4 | 85.7 | 3.5 | 91.1 | 9.0 | 86.3 | 12.6 |
| LLOQ (1 ng/mL) | LQC (2 ng/mL) | MQC (40 ng/mL) | HQC (150 ng/mL) | ||||||
| 8 | AA | 83.7 | 4.1 | 106.0 | 15.0 | 103.2 | 15.2 | 89.2 | 3.2 |
| 53 | Docosahexaenoic Acid | 114.2 | 5.6 | 111.5 | 12.2 | 94.5 | 12.2 | 85.0 | 10.8 |
| 14 | LTE4 | 109.7 | 8.4 | 112.9 | 1.7 | 90.1 | 1.7 | 92.9 | 2.5 |
| 15 | LTD4 | 112.2 | 9.9 | 85.2 | 1.7 | 93.1 | 1.7 | 112.0 | 1.8 |
| 16 | LTC4 | 114.6 | 11.4 | 91.5 | 4.9 | 93.4 | 4.9 | 114.3 | 0.2 |
| 41 | 14,15-LTC4 | 98.8 | 9.1 | 86.4 | 5.0 | 90.2 | 5.0 | 112.6 | 3.1 |
| Time (Min) | % of Aqueous Phase (A) | % of Organic Phase (B) | Flow Rate (mL/Min) |
|---|---|---|---|
| 0 | 80 | 20 | 0.37 |
| 10 | 60 | 40 | 0.37 |
| 20 | 0 | 100 | 0.37 |
| 23 | 0 | 100 | 0.37 |
| 23.1 | 80 | 20 | 0.37 |
| 25 | 80 | 20 | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhonker, Y.S.; Kanvinde, S.; Ahmad, R.; Singh, A.B.; Oupický, D.; Murry, D.J. Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites 2021, 11, 106. https://doi.org/10.3390/metabo11020106
Chhonker YS, Kanvinde S, Ahmad R, Singh AB, Oupický D, Murry DJ. Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites. 2021; 11(2):106. https://doi.org/10.3390/metabo11020106
Chicago/Turabian StyleChhonker, Yashpal S., Shrey Kanvinde, Rizwan Ahmad, Amar B. Singh, David Oupický, and Daryl J. Murry. 2021. "Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS" Metabolites 11, no. 2: 106. https://doi.org/10.3390/metabo11020106
APA StyleChhonker, Y. S., Kanvinde, S., Ahmad, R., Singh, A. B., Oupický, D., & Murry, D. J. (2021). Simultaneous Quantitation of Lipid Biomarkers for Inflammatory Bowel Disease Using LC–MS/MS. Metabolites, 11(2), 106. https://doi.org/10.3390/metabo11020106