
Combined Metabolome and Lipidome Analyses for In-Depth Characterization of Lipid Accumulation in the DHA Producing Aurantiochytrium sp. T66

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
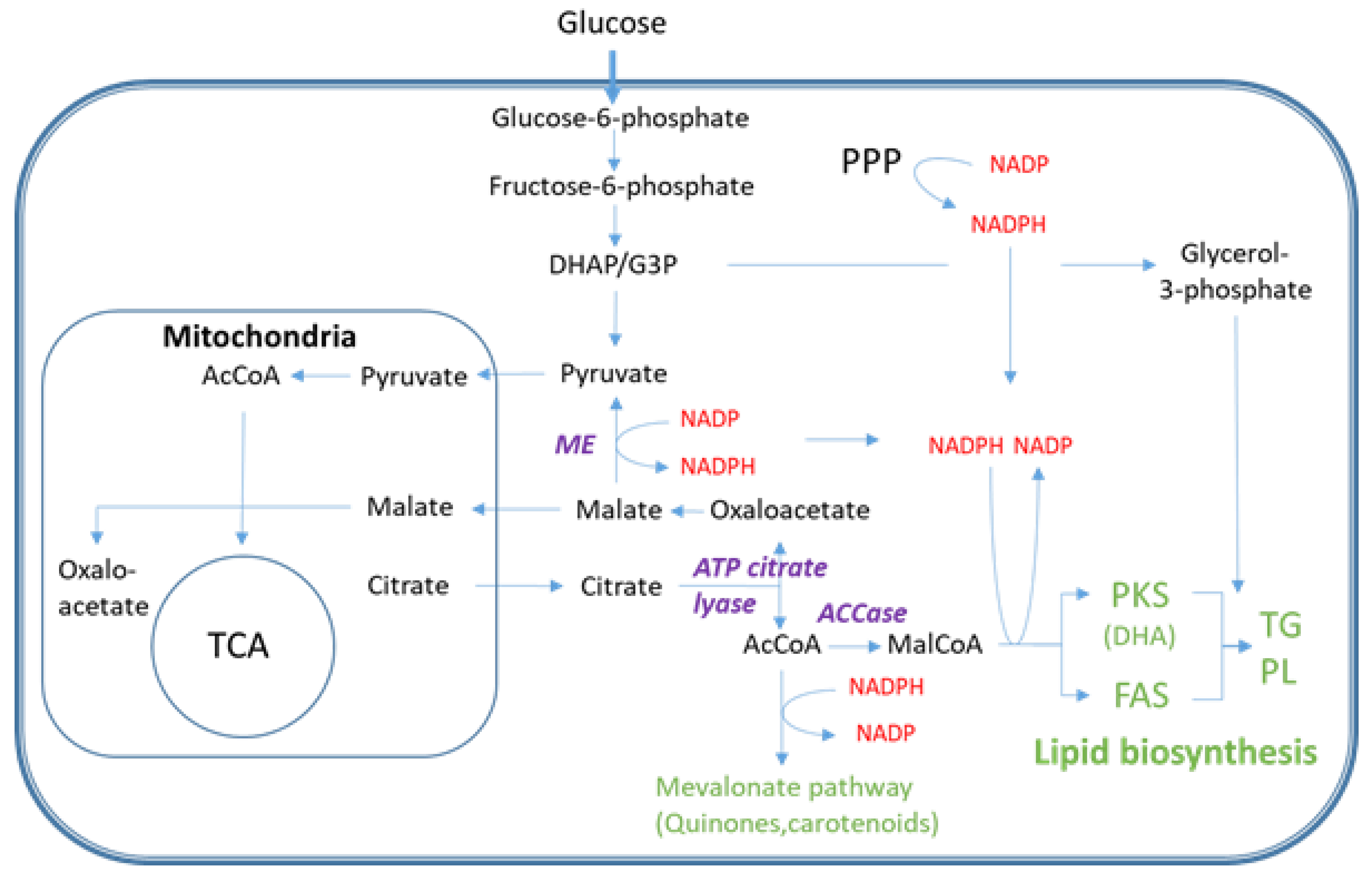
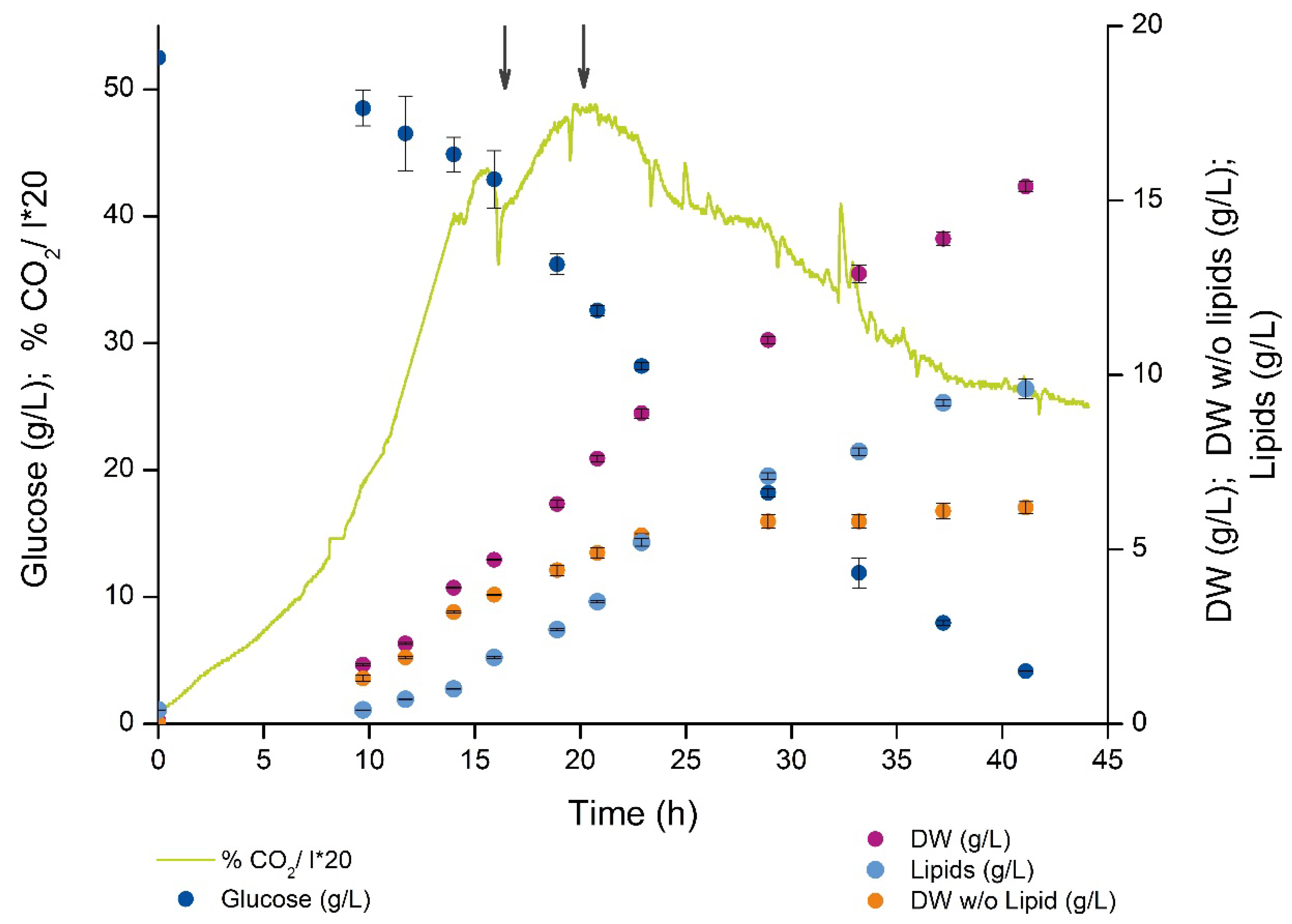
2.1. Cell Growth and Lipid Accumulation
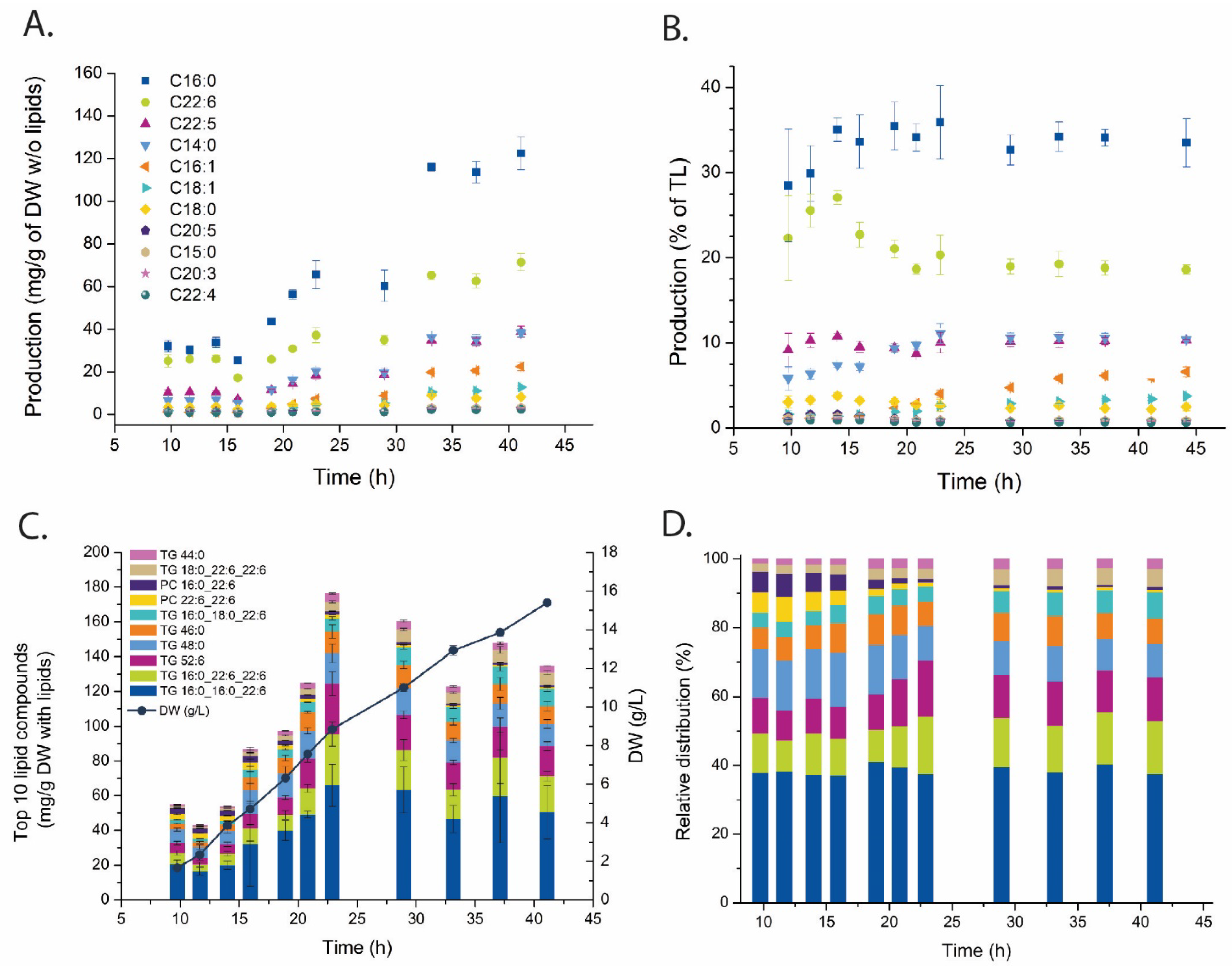
2.2. Fatty Acid and Lipid Profiling
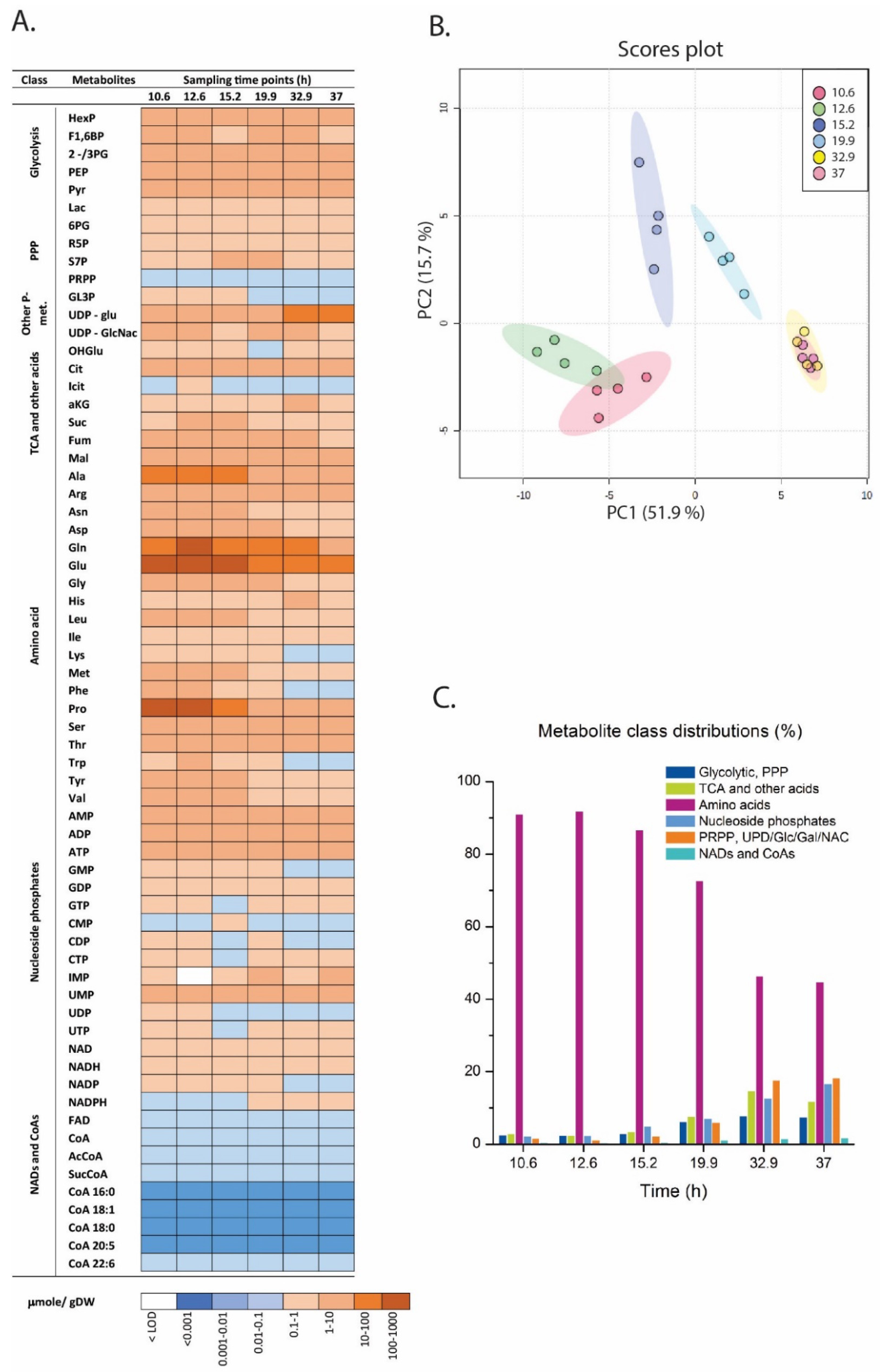
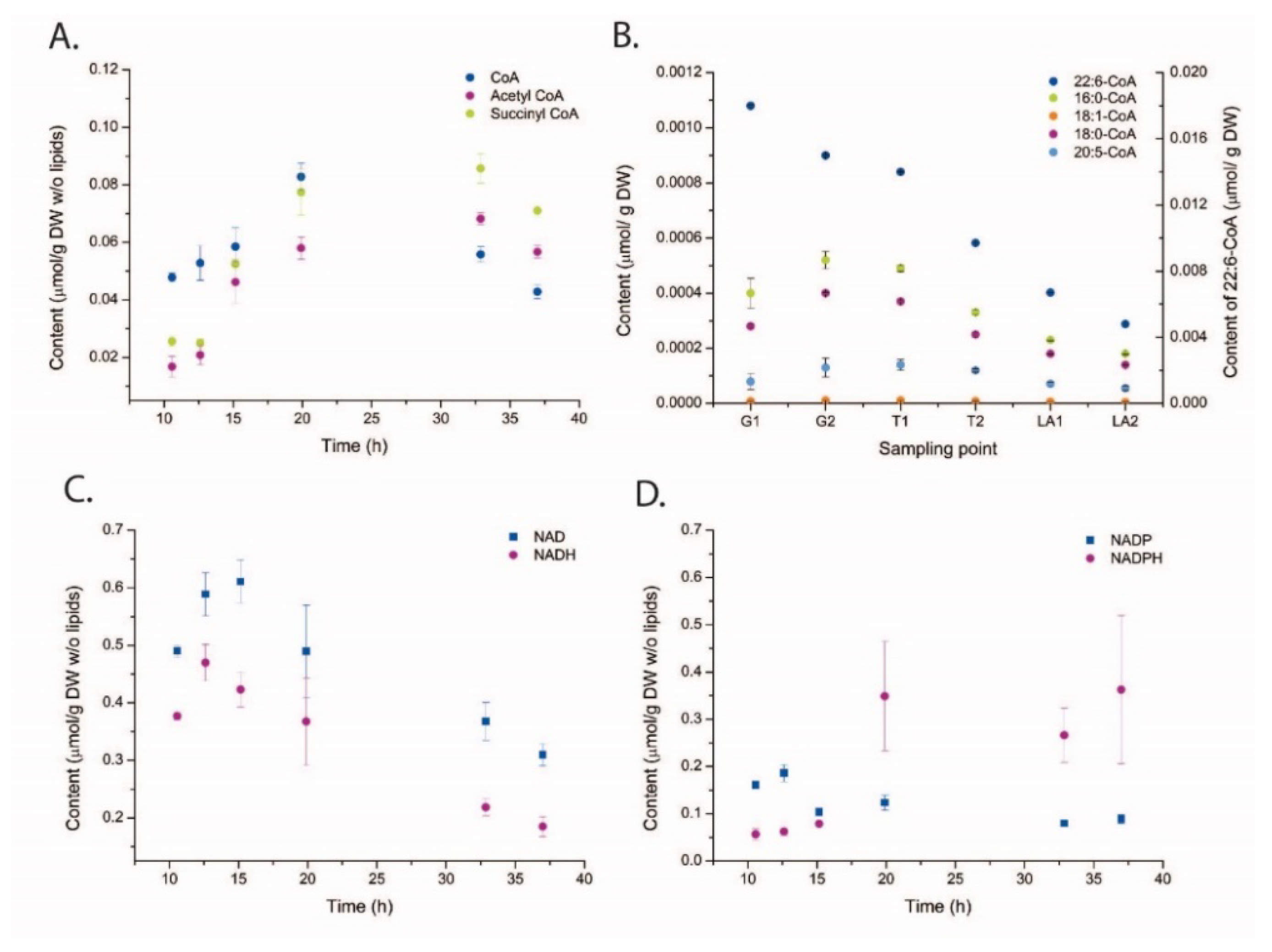
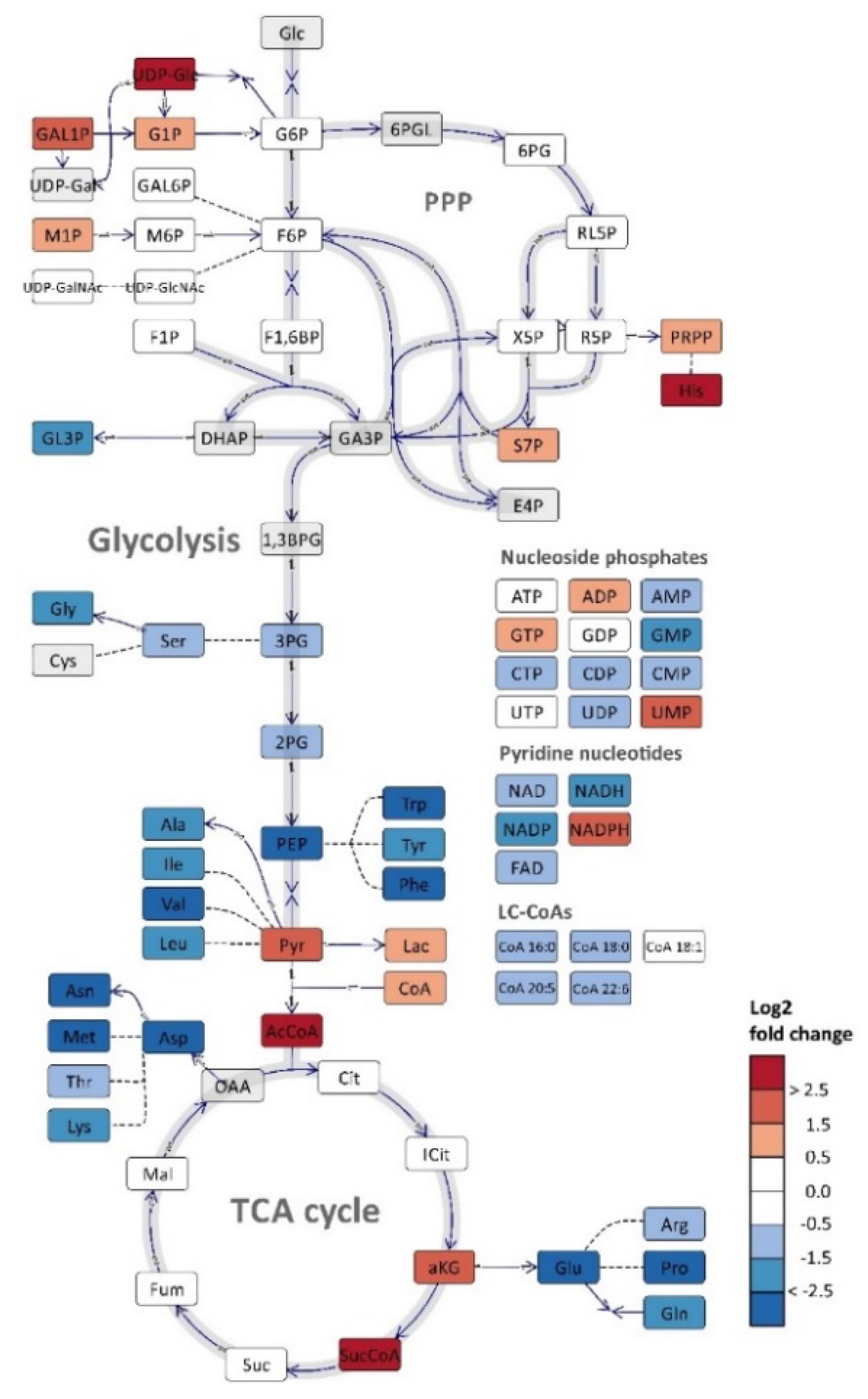
2.3. Metabolite Profiling
3. Discussion
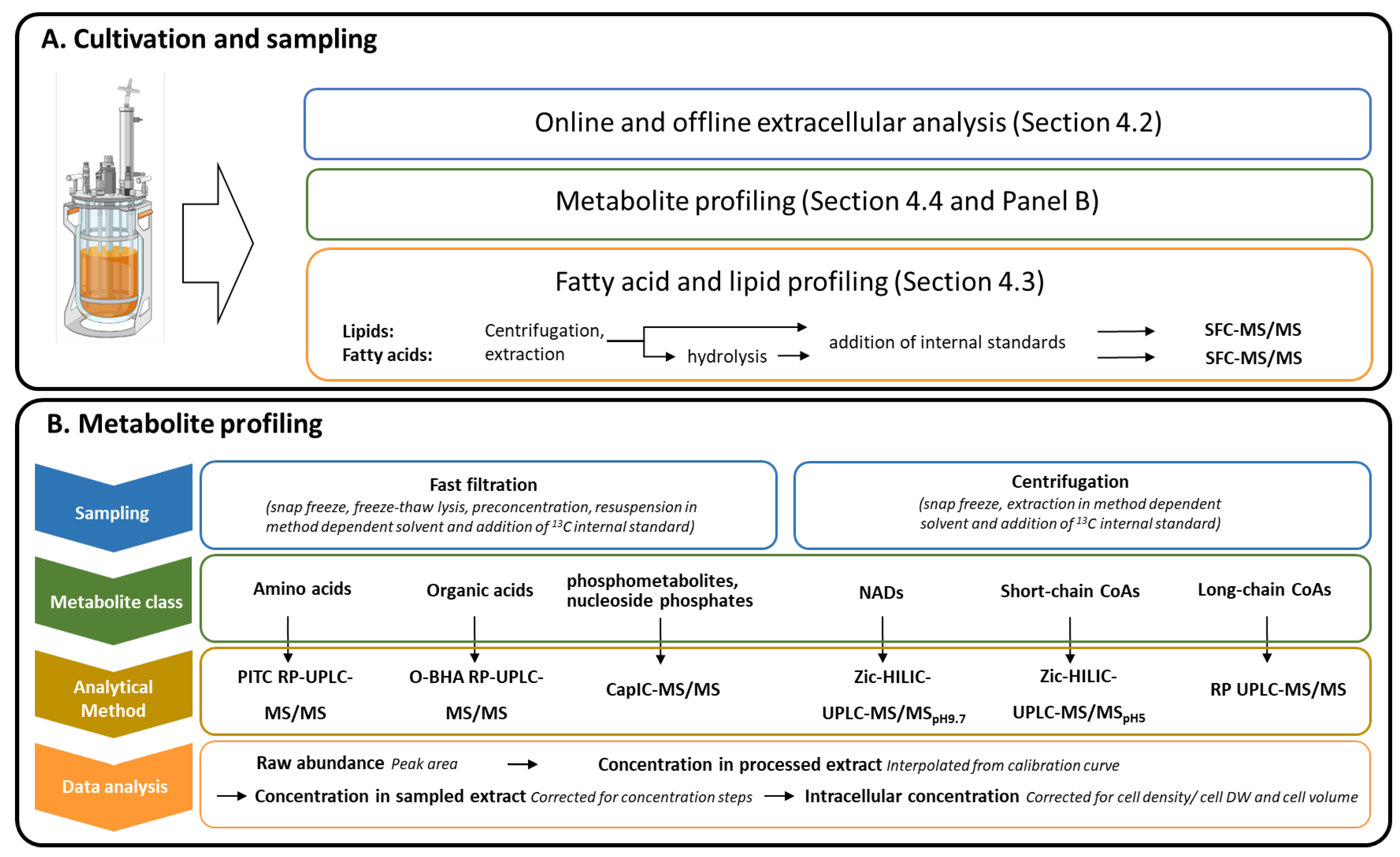
4. Materials and Methods
4.1. Cultivations
4.2. Biomass and Extracellular Analyses
4.3. Fatty Acid and Lipid Profiling
4.3.1. Lipid Extraction
4.3.2. Nontarget Lipid Analysis
4.3.3. Hydrolysis of Lipids
4.3.4. Quantitative Analysis of Fatty Acids
4.4. Metabolite Profiling
4.4.1. Extraction and Quantitation of CoA-Metabolites
4.4.2. Comments on the Optimization of Extraction and Quantitation of the NAD- and SC-CoA-Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aasen, I.M.; Ertesvåg, H.; Heggeset, T.M.B.; Liu, B.; Brautaset, T.; Vadstein, O.; Ellingsen, T.E. Thraustochytrids as production organisms for docosahexaenoic acid (DHA), squalene, and carotenoids. Appl. Microbiol. Biotechnol. 2016, 100, 4309–4321. [Google Scholar] [CrossRef] [PubMed]
- Marchan, L.F.; Chang, K.J.L.; Nichols, P.D.; Mitchell, W.J.; Polglase, J.L.; Gutierrez, T. Taxonomy, ecology and biotechnological applications of thraustochytrids: A review. Biotechnol. Adv. 2018, 36, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Colonia, B.S.O.; Pereira, G.V.D.M.; Soccol, C.R. Omega-3 microbial oils from marine thraustochytrids as a sustainable and technological solution: A review and patent landscape. Trends Food Sci. Technol. 2020, 99, 244–256. [Google Scholar] [CrossRef]
- Patel, A.; Karageorgou, D.; Rova, E.; Katapodis, P.; Rova, U.; Christakopoulos, P.; Matsakas, L. An Overview of Potential Oleaginous Microorganisms and Their Role in Biodiesel and Omega-3 Fatty Acid-Based Industries. Microorganisms 2020, 8, 434. [Google Scholar] [CrossRef]
- Morabito, C.; Bournaud, C.; Maës, C.; Schuler, M.; Cigliano, R.A.; Dellero, Y.; Maréchal, E.; Amato, A.; Rébeillé, F. The lipid metabolism in thraustochytrids. Prog. Lipid Res. 2019, 76, 18. [Google Scholar] [CrossRef]
- Heggeset, T.M.B.; Ertesvåg, H.; Liu, B.; Ellingsen, T.E.; Vadstein, O.; Aasen, I.M. Lipid and DHA-production in Aurantiochytrium sp.—Responses to nitrogen starvation and oxygen limitation revealed by analyses of production kinetics and global transcriptomes. Sci. Rep. 2019, 9, 19470. [Google Scholar] [CrossRef] [PubMed]
- Iwasaka, H.; Koyanagi, R.; Satoh, R.; Watanabe, K.; Hisata, K.; Satoh, N.; Aki, T. A Possible Trifunctional beta-Carotene Synthase Gene Identified in the Draft Genome of Aurantiochytrium sp. Strain KH105. Genes 2018, 9, 200. [Google Scholar] [CrossRef]
- Dellero, Y.; Cagnac, O.; Rose, S.; Seddiki, K.; Cussac, M.; Morabito, C.; Lupette, J.; Cigliano, R.A.; Sanseverino, W.; Kuntz, M. Proposal of a new thraustochytrid genus Hondaea gen. nov and comparison of its lipid dynamics with the closely related pseudo-cryptic genus Aurantiochytrium. Algal Res. Biomass Biofuels Bioprod. 2018, 35, 125–141. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, P.-P.; Zhang, M.; Zhu, Y.-M.; Wang, X.-P.; Luo, X.-A.; Bao, Z.-D.; Yu, L.-J. Transcriptome analysis reveals that up-regulation of the fatty acid synthase gene promotes the accumulation of docosahexaenoic acid in Schizochytrium sp S056 when glycerol is used. Algal Res. Biomass Biofuels Bioprod. 2016, 15, 83–92. [Google Scholar] [CrossRef]
- Yue, X.-H.; Chen, W.-C.; Wang, Z.-M.; Liu, P.-Y.; Li, X.-Y.; Lin, C.-B.; Lu, S.-H.; Huang, F.-H.; Wan, X. Lipid Distribution Pattern and Transcriptomic Insights Revealed the Potential Mechanism of Docosahexaenoic Acid Traffics in Schizochytrium sp. A-2. J. Agric. Food Chem. 2019, 67, 9683–9693. [Google Scholar] [CrossRef]
- Hauvermale, A.; Kuner, J.; Rosenzweig, B.; Guerra, D.; Diltz, S.; Metz, J.G. Fatty acid production in Schizochytrium sp.: Involvement of a polyunsaturated fatty acid synthase and a type I fatty acid synthase. Lipids 2006, 41, 739–747. [Google Scholar] [CrossRef]
- Liu, B.; Ertesvåg, H.; Aasen, I.M.; Vadstein, O.; Brautaset, T.; Heggeset, T.M.B. Draft genome sequence of the docosahexaenoic acid producing thraustochytrid Aurantiochytrium sp. T66. Genom. Data 2016, 8, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Qiu, X. Analysis of the biosynthetic process of fatty acids in Thraustochytrium. Biochimie 2018, 144, 108–114. [Google Scholar] [CrossRef]
- Ratledge, C. The role of malic enzyme as the provider of NADPH in oleaginous microorganisms: A reappraisal and unsolved problems. Biotechnol. Lett. 2014, 36, 1557–1568. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yang, Y.T. Combining engineering strategies and fermentation technology to enhance docosahexaenoic acid (DHA) production from an indigenous Thraustochytrium sp BM2 strain. Biochem. Eng. J. 2018, 133, 179–185. [Google Scholar] [CrossRef]
- Jakobsen, A.N.; Aasen, I.M.; Josefsen, K.D.; Strøm, A.R. Accumulation of docosahexaenoic acid-rich lipid in thraustochytrid Aurantiochytrium sp strain T66: Effects of N and P starvation and O (2) limitation. Appl. Microbiol. Biotechnol. 2008, 80, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-S.; Ji, X.-J.; Ren, L.-J.; Li, G.-L.; Sun, X.-M.; Chen, K.-Q.; Gao, S.; Huang, H. Development of a scale-up strategy for fermentative production of docosahexaenoic acid by Schizochytrium sp. Chem. Eng. Sci. 2018, 176, 600–608. [Google Scholar] [CrossRef]
- Guo, D.S.; XJ, J.; LJ, R.; GL, L.; FW, Y. Development of a real-time bioprocess monitoring method for docosahexaenoic acid production by Schizochytrium sp. Bioresour. Technol. 2016, 216, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Janthanomsuk, P.; Verduyn, C.; Chauvatcharin, S. Improved docosahexaenoic acid production in Aurantiochytrium by glucose limited pH-auxostat fed-batch cultivation. Bioresour. Technol. 2015, 196, 592–599. [Google Scholar] [CrossRef]
- Kim, K.; Kim, E.J.; Ryu, B.-G.; Park, S.; Choi, Y.-E.; Yang, J.-W. A novel fed-batch process based on the biology of Aurantiochytrium sp KRS101 for the production of biodiesel and docosahexaenoic acid. Bioresour. Technol. 2013, 135, 269–274. [Google Scholar] [CrossRef]
- Wang, Q.Z.; Ye, H.; Sen, B.; Xie, Y.; He, Y.; Park, S.; Wnag, G. Improved production of docosahexaenoic acid in batch fermentation by newly-isolated strains of Schizochytrium sp. and Thraustochytriidae sp. through bioprocess optimization. Synth. Syst. Biotechnol. 2018, 3, 121–129. [Google Scholar] [CrossRef]
- Xu, X.; Huang, C.; Xu, Z.; Xu, H.; Wang, Z.; Yu, X. The strategies to reduce cost and improve productivity in DHA production by Aurantiochytrium sp.: From biochemical to genetic respects. Appl. Microbiol. Biotechnol. 2020, 104, 9433–9447. [Google Scholar] [CrossRef]
- Sun, X.-M.; Ren, L.-J.; Zhao, Q.-Y.; Ji, X.-J.; Huang, H. Enhancement of lipid accumulation in microalgae by metabolic engineering. Biochimica Et Biophysica Acta-Mol. Cell Biol. Lipids 2019, 1864, 552–566. [Google Scholar] [CrossRef]
- Cui, G.-Z.; Ma, Z.; Liu, Y.-J.; Feng, Y.; Sun, Z.; Cheng, Y.; Song, X.; Cui, Q. Overexpression of glucose-6-phosphate dehydrogenase enhanced the polyunsaturated fatty acid composition of Aurantiochytrium sp SD116. Algal Res. Biomass Biofuels Bioprod. 2016, 19, 138–145. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Matsuda, T.; Kobayashi, T.; Ohara, J.-I.; Hamaguchi, R.; Abe, E.; Nagano, N.; Hayashi, M.; Ueda, M.; Honda, D.; et al. Versatile transformation system that is applicable to both multiple transgene expression and gene targeting for Thraustochytrids. Appl. Env. Microbiol. 2012, 78, 3193–3202. [Google Scholar] [CrossRef] [PubMed]
- Merkx-Jacques, A.; Rasmussen, H.; Muise, D.M.; Benjamin, J.J.R.; Kottwitz, H.; Tanner, K.; Milway, M.T.; Purdue, L.M.; Scaife, M.A.; Armenta, R.E.; et al. Engineering xylose metabolism in thraustochytrid T18. Biotechnol. Biofuels 2018, 11, 248. [Google Scholar] [CrossRef] [PubMed]
- Faktorová, D.; Nisbet, R.E.R.; Robledo, J.A.F.; Casacuberta, E.; Sudek, L.; Allen, A.E.; Ares, M., Jr.; Aresté, C.; Balestreri, C.; Barbrook, A.C.; et al. Genetic tool development in marine protists: Emerging model organisms for experimental cell biology. bioRxiv 2020, 71, 8239. [Google Scholar]
- Sun, H.; Chen, H.; Zang, X.; Hou, P.; Zhou, B.; Liu, Y.; Wu, F.; Cao, X.; Zhang, X. Application of the Cre/loxP Site-Specific Recombination System for Gene Transformation in Aurantiochytrium limacinum. Molecules 2015, 20, 10110–10121. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Chen, S.; Sun, X.; Hu, X.; Ji, X.; Huang, H.; Ren, L. Fermentation performance and metabolomic analysis of an engineered high-yield PUFA-producing strain of Schizochytrium sp. Bioprocess. Biosyst. Eng. 2019, 42, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.M.T.; Watanabe, K.; Okamura, Y.; Nakashimada, Y.; Aki, T. Metabolite Profile Analysis of Aurantiochytrium limacinum SR21 Grown on Acetate-based Medium for Lipid Fermentation. J. Oleo. Sci. 2019, 68, 541–549. [Google Scholar] [CrossRef]
- Yang, J.; Song, X.; Wang, L.; Cui, Q. Comprehensive Analysis of Metabolic Alterations in Schizochytrium sp. Strains with Different DHA Content. J. Chromatogr. B 2020, 1160, 122193. [Google Scholar] [CrossRef]
- Liu, L.; Wang, F.; Pei, G.; Cui, J.; Diao, J.; Lv, M.; Chen, L.; Zhang, W. Repeated fed-batch strategy and metabolomic analysis to achieve high docosahexaenoic acid productivity in Crypthecodinium cohnii. Microb. Cell Factories 2020, 19, 1–14. [Google Scholar] [CrossRef]
- Bartosova, Z.; Gonzalez, S.V.; Voigt, A.; Bruheim, P. High. throughput semi-quantitative UHPSFC-MS/MS lipid profiling and lipid class determination. J. Chromatogr. Sci. 2020. [Google Scholar] [CrossRef]
- Røst, L.M.; Thorfinnsdottir, L.B.; Kumar, K.; Fuchino, K.; Langørgen, I.E.; Bartosova, Z.; Kristiansen, K.A.; Bruheim, P. Absolute Quantification of the Central Carbon Metabolome in Eight Commonly Applied Prokaryotic and Eukaryotic Model Systems. Metabolites 2020, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Seifar, R.M.; Ras, C.; van Dam, J.C.; van Gulik, J.C.; Heijnen, J.J.; van Winden, W.A. Simultaneous quantification of free nucleotides in complex biological samples using ion pair reversed phase liquid chromatography isotope dilution tandem mass spectrometry. Anal. Biochem. 2009, 388, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Chubukov, V.; Gerosa, L.; Kochanowski, K.; Sauer, U. Coordination of microbial metabolism. Nat. Rev. Microbiol. 2014, 12, 327–340. [Google Scholar] [CrossRef]
- Huergo, L.F.; Dixon, R. The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol. Mol. Biol. Rev. 2015, 79, 419–435. [Google Scholar] [CrossRef]
- Droste, P.; Miebach, S.; Niedenführ, S.; Wiechert, W.; Nöh, K. Visualizing multi-omics data in metabolic networks with the software Omix-A case study. Biosystems 2011, 105, 154–161. [Google Scholar] [CrossRef]
- Bruins, A.P. Mechanistic aspects of electrospray ionization. J. Chromatogr. A 1998, 794, 345–357. [Google Scholar] [CrossRef]
- Zhou, W.L.; Yang, S.; Wang, P.G. Matrix effects and application of matrix effect factor. Bioanalysis 2017, 9, 1839–1844. [Google Scholar] [CrossRef]
- Kvitvang, H.F.N.; Kristiansen, K.A.; Bruheim, P. Assessment of capillary anion exchange ion chromatography tandem mass spectrometry for the quantitative profiling of the phosphometabolome and organic acids in biological extracts. J. Chromatogr. A 2014, 1370, 70–79. [Google Scholar] [CrossRef]
- Stafsnes, M.H.; Røst, L.M.; Bruheim, P. Improved phosphometabolome profiling applying isotope dilution strategy and capillary ion chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1083, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Bruheim, P.; Sletta, H.; Bibb, M.J.; White, J.; Levine, D.W. High-yield actinorhodin production in fed-batch culture by a Streptomyces lividans strain overexpressing the pathway-specific activator gene actII-ORF4. J. Ind. Microbiol. Biotechnol. 2002, 28, 103–111. [Google Scholar] [CrossRef]
- Pomraning, K.R.; Wei, S.; Karagiosis, S.A.; Kim, Y.-M.; Dohnalkova, A.C.; Arey, B.W.; Bredeweg, E.L.; Orr, G.; Metz, T.O.; Baker, S.E. Comprehensive Metabolomic, Lipidomic and Microscopic Profiling of Yarrowia lipolytica during Lipid Accumulation Identifies Targets for Increased Lipogenesis. PLoS ONE 2015, 10, e0123188. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Li, X.; Liu, L.; Liu, J.; Wang, F.; Chen, L.; Zhang, W. De novo transcriptomic and metabolomic analysis of docsahexaenoic acid (DHA)-producing Crypthecodinium cohnii during fed-batch fermentation. Algal Res. 2017, 26, 380–391. [Google Scholar] [CrossRef]
- Shen, Y.; Fatemeh, T.; Tang, L.; Cai, Z. Quantitative metabolic network profiling of Escherichia coli: An. overview of analytical methods for measurement of intracellular metabolites. Trac-Trends Anal. Chem. 2016, 75, 141–150. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Røst, L.M.; Shafaei, A.; Fuchino, K.; Bruheim, P. Zwitterionic HILIC tandem mass spectrometry with isotope dilution for rapid, sensitive and robust quantification of pyridine nucleotides in biological extracts. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1144, 122078. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Minkler, P.E.; Kerner, J.; Ingalls, S.T.; Hoppel, C.L. Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal. Biochem. 2008, 376, 275–276. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartosova, Z.; Ertesvåg, H.; Nyfløt, E.L.; Kämpe, K.; Aasen, I.M.; Bruheim, P. Combined Metabolome and Lipidome Analyses for In-Depth Characterization of Lipid Accumulation in the DHA Producing Aurantiochytrium sp. T66. Metabolites 2021, 11, 135. https://doi.org/10.3390/metabo11030135
Bartosova Z, Ertesvåg H, Nyfløt EL, Kämpe K, Aasen IM, Bruheim P. Combined Metabolome and Lipidome Analyses for In-Depth Characterization of Lipid Accumulation in the DHA Producing Aurantiochytrium sp. T66. Metabolites. 2021; 11(3):135. https://doi.org/10.3390/metabo11030135
Chicago/Turabian StyleBartosova, Zdenka, Helga Ertesvåg, Eirin Lishaugen Nyfløt, Kristoffer Kämpe, Inga Marie Aasen, and Per Bruheim. 2021. "Combined Metabolome and Lipidome Analyses for In-Depth Characterization of Lipid Accumulation in the DHA Producing Aurantiochytrium sp. T66" Metabolites 11, no. 3: 135. https://doi.org/10.3390/metabo11030135
APA StyleBartosova, Z., Ertesvåg, H., Nyfløt, E. L., Kämpe, K., Aasen, I. M., & Bruheim, P. (2021). Combined Metabolome and Lipidome Analyses for In-Depth Characterization of Lipid Accumulation in the DHA Producing Aurantiochytrium sp. T66. Metabolites, 11(3), 135. https://doi.org/10.3390/metabo11030135