
Hepatitis C Virus Uses Host Lipids to Its Own Advantage


{kind=link}
{kind=link}
{kind=link}
Abstract
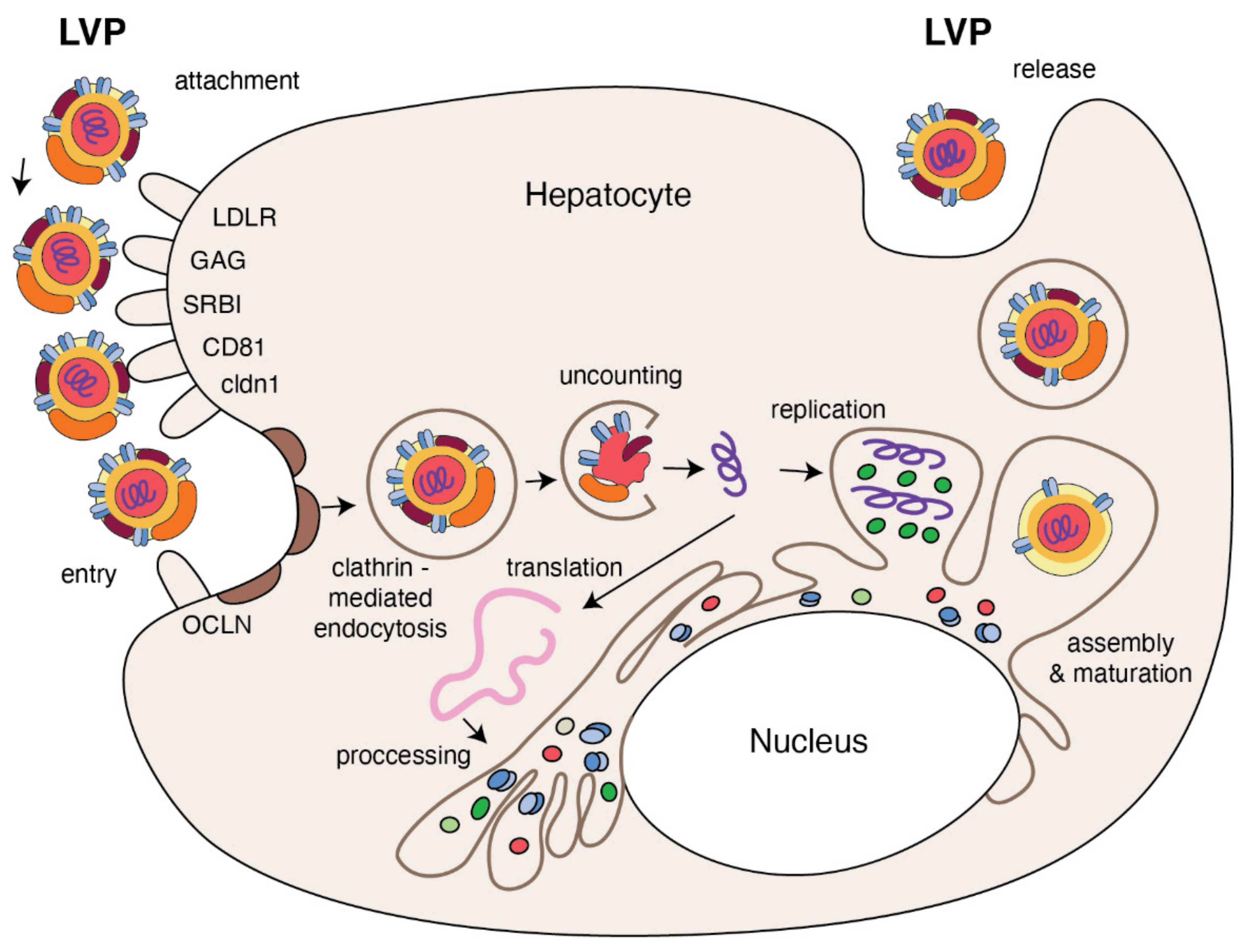
:1. Introduction
2. Lipid Phenotype of Chronic HCV Patients
3. Resemblance between HCV Particles and Lipoproteins
4. Lipid-Dependent HCV Entry
5. Host Factors Contribution to the Efficient HCV Replication
6. HCV Hijacks the VLDL Secretory Pathway for Progeny Formation
7. Conclusions
Funding
Conflicts of Interest
References
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Houghton, M. The long and winding road leading to the identification of the hepatitis C virus. J. Hepatol. 2009, 51, 939–948. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, M.; Andreo, U.; Chung, H.Y.; Espiritu, C.; Branch, A.D.; Silva, J.M.; Rice, C.M. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature 2015, 524, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roudot-Thoraval, F. Epidemiology of hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101596. [Google Scholar] [CrossRef] [PubMed]
- Sagnelli, E.; Macera, M.; Russo, A.; Coppola, N.; Sagnelli, C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection 2020, 48, 7–17. [Google Scholar] [CrossRef]
- Bamber, M.; Murray, A.K.; Weller, I.V.; Morelli, A.; Scheuer, P.J.; Thomas, H.C.; Sherlock, S. Clinical and histological features of a group of patients with sporadic non-A, non-B hepatitis. J. Clin. Pathol. 1981, 34, 1175–1180. [Google Scholar] [CrossRef]
- Fabris, C.; Federico, E.; Soardo, G.; Falleti, E.; Pirisi, M. Blood lipids of patients with chronic hepatitis: Differences related to viral etiology. Clin. Chim. Acta 1997, 261, 159–165. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Köchel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef]
- André, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Bréchot, C.; Paranhos-Baccalà, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; von Schaewen, M.; Ploss, A. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 2014, 16, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. Hepatitis C Virus Replication. Adv. Exp. Med. Biol. 2017, 997, 199–209. [Google Scholar] [CrossRef]
- Sharma, P.; Balan, V.; Hernandez, J.; Rosati, M.; Williams, J.; Rodriguez-Luna, H.; Schwartz, J.; Harrison, E.; Anderson, M.; Byrne, T.; et al. Hepatic steatosis in hepatitis C virus genotype 3 infection: Does it correlate with body mass index, fibrosis, and HCV risk factors? Dig. Dis. Sci. 2004, 49, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Thomopoulos, K.C.; Arvaniti, V.; Tsamantas, A.C.; Dimitropoulou, D.; Gogos, C.A.; Siagris, D.; Theocharis, G.J.; Labropoulou-Karatza, C. Prevalence of liver steatosis in patients with chronic hepatitis B: A study of associated factors and of relationship with fibrosis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Sanyal, A.J. Hepatitis C virus, steatosis and lipid abnormalities: Clinical and pathogenic data. Liver Int. 2009, 29 (Suppl. S2), 26–37. [Google Scholar] [CrossRef]
- Piodi, A.; Chouteau, P.; Lerat, H.; Hézode, C.; Pawlotsky, J.M. Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology 2008, 48, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.; Herker, E.; Farese, R.V.; Ott, M. Hepatitis C virus core protein decreases lipid droplet turnover: A mechanism for core-induced steatosis. J. Biol. Chem. 2011, 286, 42615–42625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.L.; Yeh, C.T.; Chen, J.C.; Huang, C.C.; Lin, S.M.; Sheen, I.S.; Tai, D.I.; Chu, C.M.; Lin, W.P.; Chang, M.Y.; et al. Altered expression patterns of lipid metabolism genes in an animal model of HCV core-related, nonobese, modest hepatic steatosis. BMC Genom. 2008, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Lerat, H.; Kammoun, H.L.; Hainault, I.; Mérour, E.; Higgs, M.R.; Callens, C.; Lemon, S.M.; Foufelle, F.; Pawlotsky, J.M. Hepatitis C virus proteins induce lipogenesis and defective triglyceride secretion in transgenic mice. J. Biol. Chem. 2009, 284, 33466–33474. [Google Scholar] [CrossRef] [Green Version]
- Enjoji, M.; Kohjima, M.; Kotoh, K.; Nakamuta, M. Metabolic disorders and steatosis in patients with chronic hepatitis C: Metabolic strategies for antiviral treatments. Int. J. Hepatol. 2012, 2012, 264017. [Google Scholar] [CrossRef] [PubMed]
- Domitrovich, A.M.; Felmlee, D.J.; Siddiqui, A. Hepatitis C virus nonstructural proteins inhibit apolipoprotein B100 secretion. J. Biol. Chem. 2005, 280, 39802–39808. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, S.; Realdon, S.; Iqbal, J.; Gerotto, M.; Dal Pero, F.; Bortoletto, G.; Marcolongo, M.; Vario, A.; Datz, C.; Hussain, M.M.; et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology 2006, 130, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Honda, A.; Ikegami, T.; Saitoh, Y.; Hirayama, T.; Hara, T.; Doy, M.; Matsuzaki, Y. Hepatitis C virus infection causes hypolipidemia regardless of hepatic damage or nutritional state: An epidemiological survey of a large Japanese cohort. Hepatol. Res. 2011, 41, 530–541. [Google Scholar] [CrossRef]
- Sidorkiewicz, M.; Grek-Kowalinska, M.; Piekarska, A. The Correlation between miR-122 and Lipoprotein Lipase Expression in Chronic Hepatitis C Patients. Can. J. Gastroenterol. Hepatol. 2018, 2018, 6348948. [Google Scholar] [CrossRef]
- Lambert, J.E.; Bain, V.G.; Ryan, E.A.; Thomson, A.B.; Clandinin, M.T. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology 2013, 57, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chrétien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2002, 16, 185–194. [Google Scholar] [CrossRef]
- Serfaty, L.; Andreani, T.; Giral, P.; Carbonell, N.; Chazouillères, O.; Poupon, R. Hepatitis C virus induced hypobetalipoproteinemia: A possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 2001, 34, 428–434. [Google Scholar] [CrossRef]
- Corey, K.E.; Kane, E.; Munroe, C.; Barlow, L.L.; Zheng, H.; Chung, R.T. Hepatitis C virus infection and its clearance alter circulating lipids: Implications for long-term follow-up. Hepatology 2009, 50, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Chida, T.; Kawata, K.; Ohta, K.; Matsunaga, E.; Ito, J.; Shimoyama, S.; Yamazaki, S.; Noritake, H.; Suzuki, T.; Suda, T.; et al. Rapid Changes in Serum Lipid Profiles during Combination Therapy with Daclatasvir and Asunaprevir in Patients Infected with Hepatitis C Virus Genotype 1b. Gut Liver 2018, 12, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Corey, K.E.; Mendez-Navarro, J.; Barlow, L.L.; Patwardhan, V.; Zheng, H.; Kim, A.Y.; Lauer, G.M.; Chung, R.T. Acute hepatitis C infection lowers serum lipid levels. J. Viral Hepat. 2011, 18, e366–e371. [Google Scholar] [CrossRef] [Green Version]
- Lavillette, D.; Pécheur, E.I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765. [Google Scholar] [CrossRef] [Green Version]
- Pumeechockchai, W.; Bevitt, D.; Agarwal, K.; Petropoulou, T.; Langer, B.C.; Belohradsky, B.; Bassendine, M.F.; Toms, G.L. Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: Implications for virus clearance by antibody. J. Med. Virol. 2002, 68, 335–342. [Google Scholar] [CrossRef]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.U.; Bassendine, M.F.; Martin, C.; Lowther, D.; Purcell, P.J.; King, B.J.; Neely, D.; Toms, G.L. Characterization of hepatitis C RNA-containing particles from human liver by density and size. J. Gen. Virol. 2008, 89, 2507–2517. [Google Scholar] [CrossRef]
- Ramasamy, I. Recent advances in physiological lipoprotein metabolism. Clin. Chem. Lab. Med. 2014, 52, 1695–1727. [Google Scholar] [CrossRef]
- Alonzi, T.; Mancone, C.; Amicone, L.; Tripodi, M. Elucidation of lipoprotein particles structure by proteomic analysis. Expert Rev. Proteom. 2008, 5, 91–104. [Google Scholar] [CrossRef]
- Bridge, S.H.; Sheridan, D.A.; Felmlee, D.J.; Nielsen, S.U.; Thomas, H.C.; Taylor-Robinson, S.D.; Neely, R.D.; Toms, G.L.; Bassendine, M.F. Insulin resistance and low-density apolipoprotein B-associated lipoviral particles in hepatitis C virus genotype 1 infection. Gut 2011, 60, 680–687. [Google Scholar] [CrossRef]
- Sundaram, M.; Yao, Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr. Metab. 2010, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Packard, C.J.; Munro, A.; Lorimer, A.R.; Gotto, A.M.; Shepherd, J. Metabolism of apolipoprotein B in large triglyceride-rich very low density lipoproteins of normal and hypertriglyceridemic subjects. J. Clin. Investig. 1984, 74, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- Scholtes, C.; Ramière, C.; Rainteau, D.; Perrin-Cocon, L.; Wolf, C.; Humbert, L.; Carreras, M.; Guironnet-Paquet, A.; Zoulim, F.; Bartenschlager, R.; et al. High plasma level of nucleocapsid-free envelope glycoprotein-positive lipoproteins in hepatitis C patients. Hepatology 2012, 56, 39–48. [Google Scholar] [CrossRef]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef] [Green Version]
- Maillard, P.; Krawczynski, K.; Nitkiewicz, J.; Bronnert, C.; Sidorkiewicz, M.; Gounon, P.; Dubuisson, J.; Faure, G.; Crainic, R.; Budkowska, A. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J. Virol. 2001, 75, 8240–8250. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, K.; Mesalam, A.A.; Leroux-Roels, G.; Meuleman, P. Impact of lipids and lipoproteins on hepatitis C virus infection and virus neutralization. World J. Gastroenterol. 2014, 20, 15975–15991. [Google Scholar] [CrossRef] [Green Version]
- Piver, E.; Boyer, A.; Gaillard, J.; Bull, A.; Beaumont, E.; Roingeard, P.; Meunier, J.C. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut 2017, 66, 1487–1495. [Google Scholar] [CrossRef]
- Podevin, P.; Carpentier, A.; Pène, V.; Aoudjehane, L.; Carrière, M.; Zaïdi, S.; Hernandez, C.; Calle, V.; Méritet, J.F.; Scatton, O.; et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 2010, 139, 1355–1364. [Google Scholar] [CrossRef]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [Green Version]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Meex, S.J.; Andreo, U.; Sparks, J.D.; Fisher, E.A. Huh-7 or HepG2 cells: Which is the better model for studying human apolipoprotein-B100 assembly and secretion? J. Lipid Res. 2011, 52, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLoS ONE 2014, 9, e95550. [Google Scholar] [CrossRef]
- Jiang, J.; Wu, X.; Tang, H.; Luo, G. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS ONE 2013, 8, e67982. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Martinez, P.; Séron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.M.; Huang, H.; Ye, J.; Gale, M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, B.; Banerjee, A.; Meyer, K.; Ray, R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 2011, 54, 1149–1156. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Nelson, J.K.; Maspero, E.; Marques, A.R.; Scheer, L.; Polo, S.; Zelcer, N. The LXR-IDOL axis defines a clathrin-, caveolae-, and dynamin-independent endocytic route for LDLR internalization and lysosomal degradation. J. Lipid Res. 2013, 54, 2174–2184. [Google Scholar] [CrossRef] [Green Version]
- Albecka, A.; Belouzard, S.; Op de Beeck, A.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Tercé, F.; Duverlie, G.; Rouillé, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Monazahian, M.; Böhme, I.; Bonk, S.; Koch, A.; Scholz, C.; Grethe, S.; Thomssen, R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 1999, 57, 223–229. [Google Scholar] [CrossRef]
- Sheridan, D.A.; Price, D.A.; Schmid, M.L.; Toms, G.L.; Donaldson, P.; Neely, D.; Bassendine, M.F. Apolipoprotein B-associated cholesterol is a determinant of treatment outcome in patients with chronic hepatitis C virus infection receiving anti-viral agents interferon-alpha and ribavirin. Aliment Pharmacol. Ther. 2009, 29, 1282–1290. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Dao Thi, V.L.; Fresquet, J.; Guérin, M.; Julia, Z.; Verney, G.; Durantel, D.; Zoulim, F.; Lavillette, D.; Cosset, F.L.; et al. Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog. 2009, 5, e1000310. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.N.; Turek, M.; Xiao, F.; Thi, V.L.; Guérin, M.; Fofana, I.; Bachellier, P.; Thompson, J.; Delang, L.; Neyts, J.; et al. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 2013, 57, 492–504. [Google Scholar] [CrossRef]
- Maillard, P.; Huby, T.; Andréo, U.; Moreau, M.; Chapman, J.; Budkowska, A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins. FASEB J. 2006, 20, 735–737. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Cun, W. The Role of ApoE in HCV Infection and Comorbidity. Int. J. Mol. Sci. 2019, 20, 2037. [Google Scholar] [CrossRef] [Green Version]
- Zona, L.; Turek, M.; Baumert, T.F.; Zeisel, M.B. Hepatitis C virus internalization. Virologie 2013, 17, 401–413. [Google Scholar] [CrossRef]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Baktash, Y.; Madhav, A.; Coller, K.E.; Randall, G. Single Particle Imaging of Polarized Hepatoma Organoids upon Hepatitis C Virus Infection Reveals an Ordered and Sequential Entry Process. Cell Host Microbe 2018, 23, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreux, M.; Boson, B.; Ricard-Blum, S.; Molle, J.; Lavillette, D.; Bartosch, B.; Pécheur, E.I.; Cosset, F.L. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 2007, 282, 32357–32369. [Google Scholar] [CrossRef] [Green Version]
- Niepmann, M.; Gerresheim, G.K. Hepatitis C Virus Translation Regulation. Int. J. Mol. Sci. 2020, 21, 2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011, 39, 7716–7729. [Google Scholar] [CrossRef] [Green Version]
- Chahal, J.; Gebert, L.F.R.; Gan, H.H.; Camacho, E.; Gunsalus, K.C.; MacRae, I.J.; Sagan, S.M. miR-122 and Ago interactions with the HCV genome alter the structure of the viral 5′ terminus. Nucleic Acids Res. 2019, 47, 5307–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gale, M.; Keller, B.C.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 2005, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mingorance, L.; Castro, V.; Ávila-Pérez, G.; Calvo, G.; Rodriguez, M.J.; Carrascosa, J.L.; Pérez-Del-Pulgar, S.; Forns, X.; Gastaminza, P. Host phosphatidic acid phosphatase lipin1 is rate limiting for functional hepatitis C virus replicase complex formation. PLoS Pathog. 2018, 14, e1007284. [Google Scholar] [CrossRef]
- Stoeck, I.K.; Lee, J.Y.; Tabata, K.; Romero-Brey, I.; Paul, D.; Schult, P.; Lohmann, V.; Kaderali, L.; Bartenschlager, R. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, A.; Reghellin, V.; Donnici, L.; Fenu, S.; Alvarez, R.; Baruffa, C.; Peri, F.; Pagani, M.; Abrignani, S.; Neddermann, P.; et al. Metabolism of phosphatidylinositol 4-kinase IIIα-dependent PI4P Is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 2012, 8, e1002576. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.T.; Lee, K.J.; Aizaki, H.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 2003, 77, 4160–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, N.J.; Lee, C.; Pang, P.S.; Pham, E.A.; Fram, B.; Nguyen, K.; Xiong, A.; Sklan, E.H.; Elazar, M.; Koytak, E.S.; et al. Phosphatidylinositol 4,5-bisphosphate is an HCV NS5A ligand and mediates replication of the viral genome. Gastroenterology 2015, 148, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tai, A.W. Nir2 Is an Effector of VAPs Necessary for Efficient Hepatitis C Virus Replication and Phosphatidylinositol 4-Phosphate Enrichment at the Viral Replication Organelle. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Harak, C.; Meyrath, M.; Romero-Brey, I.; Schenk, C.; Gondeau, C.; Schult, P.; Esser-Nobis, K.; Saeed, M.; Neddermann, P.; Schnitzler, P.; et al. Tuning a cellular lipid kinase activity adapts hepatitis C virus to replication in cell culture. Nat. Microbiol. 2016, 2, 16247. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schöbel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Leu, G.Z.; Lin, T.Y.; Hsu, J.T. Anti-HCV activities of selective polyunsaturated fatty acids. Biochem. Biophys. Res. Commun. 2004, 318, 275–280. [Google Scholar] [CrossRef]
- Lee, J.Y.; Cortese, M.; Haselmann, U.; Tabata, K.; Romero-Brey, I.; Funaya, C.; Schieber, N.L.; Qiang, Y.; Bartenschlager, M.; Kallis, S.; et al. Spatiotemporal Coupling of the Hepatitis C Virus Replication Cycle by Creating a Lipid Droplet- Proximal Membranous Replication Compartment. Cell Rep. 2019, 27, 3602–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Sasaki, E.; Kakinuma, C.; Yano, T.; Miura, S.; Ezaki, O. Increased very low density lipoprotein secretion and gonadal fat mass in mice overexpressing liver DGAT1. J. Biol. Chem. 2005, 280, 21506–21514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. USA 1997, 94, 1200–1205. [Google Scholar] [CrossRef] [Green Version]
- Klein, K.C.; Dellos, S.R.; Lingappa, J.R. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell-free system. J. Virol. 2005, 79, 6814–6826. [Google Scholar] [CrossRef] [Green Version]
- Shi, G.; Ando, T.; Suzuki, R.; Matsuda, M.; Nakashima, K.; Ito, M.; Omatsu, T.; Oba, M.; Ochiai, H.; Kato, T.; et al. Involvement of the 3′ Untranslated Region in Encapsidation of the Hepatitis C Virus. PLoS Pathog. 2016, 12, e1005441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; et al. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [Green Version]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef]
- Icard, V.; Diaz, O.; Scholtes, C.; Perrin-Cocon, L.; Ramière, C.; Bartenschlager, R.; Penin, F.; Lotteau, V.; André, P. Secretion of hepatitis C virus envelope glycoproteins depends on assembly of apolipoprotein B positive lipoproteins. PLoS ONE 2009, 4, e4233. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Acosta, E.G.; Stoeck, I.K.; Long, G.; Hiet, M.S.; Mueller, B.; Fackler, O.T.; Kallis, S.; Bartenschlager, R. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J. Virol. 2014, 88, 12422–12437. [Google Scholar] [CrossRef] [Green Version]
- Hueging, K.; Doepke, M.; Vieyres, G.; Bankwitz, D.; Frentzen, A.; Doerrbecker, J.; Gumz, F.; Haid, S.; Wölk, B.; Kaderali, L.; et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J. Virol. 2014, 88, 1433–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, A.; Dumans, A.; Beaumont, E.; Etienne, L.; Roingeard, P.; Meunier, J.C. The association of hepatitis C virus glycoproteins with apolipoproteins E and B early in assembly is conserved in lipoviral particles. J. Biol. Chem. 2014, 289, 18904–18913. [Google Scholar] [CrossRef] [Green Version]
- Gondar, V.; Molina-Jiménez, F.; Hishiki, T.; García-Buey, L.; Koutsoudakis, G.; Shimotohno, K.; Benedicto, I.; Majano, P.L. Apolipoprotein E, but Not Apolipoprotein B, Is Essential for Efficient Cell-to-Cell Transmission of Hepatitis C Virus. J. Virol. 2015, 89, 9962–9973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, T.; Wada, M.; Nakamura, S.; Ono, C.; Shiokawa, M.; Yamamoto, S.; Motomura, T.; Okamoto, T.; Okuzaki, D.; Yamamoto, M.; et al. Amphipathic α-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLoS Pathog. 2014, 10, e1004534. [Google Scholar] [CrossRef]
- Fukuhara, T.; Tamura, T.; Ono, C.; Shiokawa, M.; Mori, H.; Uemura, K.; Yamamoto, S.; Kurihara, T.; Okamoto, T.; Suzuki, R.; et al. Host-derived apolipoproteins play comparable roles with viral secretory proteins Erns and NS1 in the infectious particle formation of Flaviviridae. PLoS Pathog. 2017, 13, e1006475. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef]
- Tiwari, S.; Siddiqi, S.A. Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosset, F.L.; Mialon, C.; Boson, B.; Granier, C.; Denolly, S. HCV Interplay with Lipoproteins: Inside or Outside the Cells? Viruses 2020, 12, 434. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, O.; Delers, F.; Maynard, M.; Demignot, S.; Zoulim, F.; Chambaz, J.; Trépo, C.; Lotteau, V.; André, P. Preferential association of Hepatitis C virus with apolipoprotein B48-containing lipoproteins. J. Gen. Virol. 2006, 87, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Denolly, S.; Granier, C.; Fontaine, N.; Pozzetto, B.; Bourlet, T.; Guérin, M.; Cosset, F.L. A serum protein factor mediates maturation and apoB-association of HCV particles in the extracellular milieu. J. Hepatol. 2019, 70, 626–638. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorkiewicz, M. Hepatitis C Virus Uses Host Lipids to Its Own Advantage. Metabolites 2021, 11, 273. https://doi.org/10.3390/metabo11050273
Sidorkiewicz M. Hepatitis C Virus Uses Host Lipids to Its Own Advantage. Metabolites. 2021; 11(5):273. https://doi.org/10.3390/metabo11050273
Chicago/Turabian StyleSidorkiewicz, Malgorzata. 2021. "Hepatitis C Virus Uses Host Lipids to Its Own Advantage" Metabolites 11, no. 5: 273. https://doi.org/10.3390/metabo11050273
APA StyleSidorkiewicz, M. (2021). Hepatitis C Virus Uses Host Lipids to Its Own Advantage. Metabolites, 11(5), 273. https://doi.org/10.3390/metabo11050273