
Asparagine: A Metabolite to Be Targeted in Cancers

Abstract
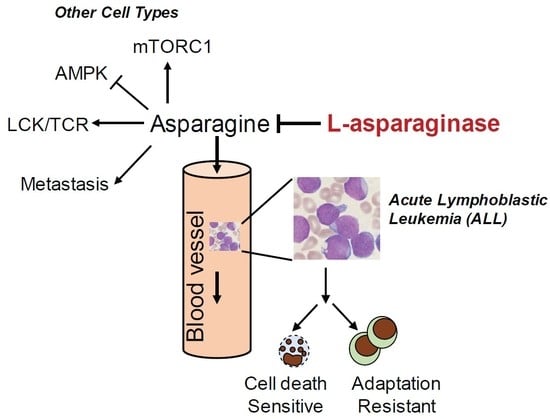
:
1. Introduction
2. Asparagine and L-Asparaginase in Acute Lymphoblastic Leukemia (ALL)
2.1. History of L-Asparaginase
2.2. Clinical Responses to L-Asparaginase Treatment in ALL Patient
2.3. Mechanism of Resistance to L-Asparaginase Treatment in ALL
3. The Role of Asparagine in Other Types of Cancer
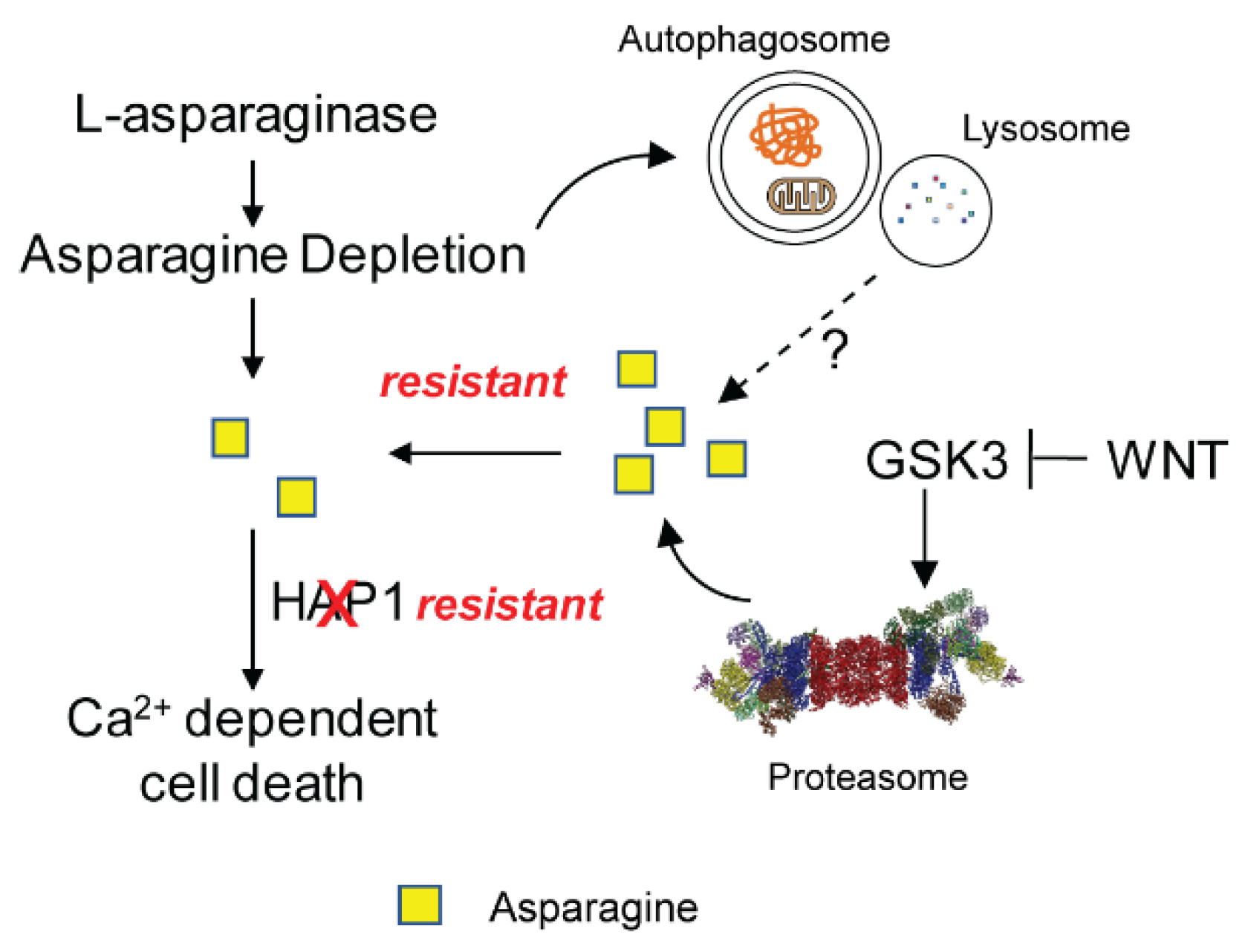
3.1. Asparagine in Promoting Solid Tumor Progression

{kind=link}
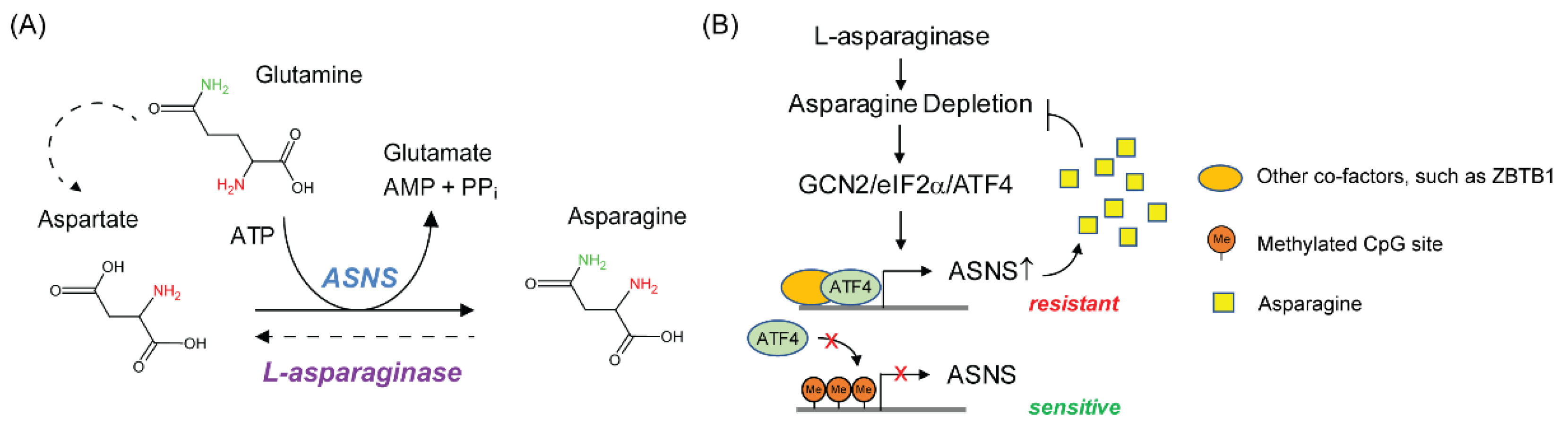{kind=link}
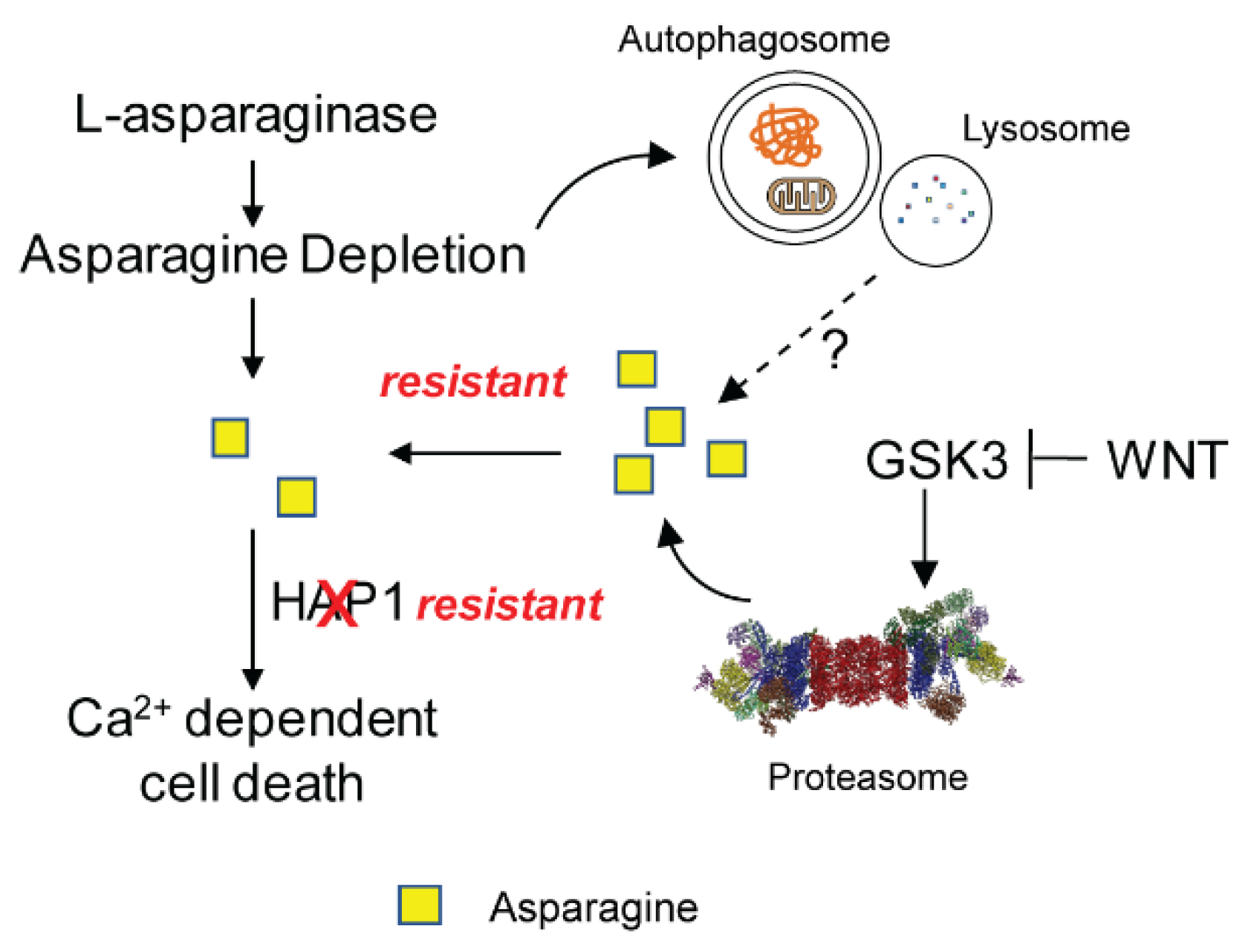{kind=link}
{kind=link}
| References | Biological Processes | Functions |
|---|---|---|
| Zhang J, [71] | Glutamine starvation | Suppresses ER stress and apoptosis |
| Pavlova NN, [25] | Glutamine starvation | Supports GLUL expression and glutamine biosynthesis |
| Gwinn DM, [75] | KRAS-driven lung cancer | NRF2-dependent de novo biosynthesis to support tumor cell growth |
| LeBoeuf SE, [76] | KRAS-driven lung cancer | Demand for uptake to mitigate NRF2-dependent glutamate export |
| Linares JF, [77] | Prostate cancer | Secreted by CAFs to support tumor cell growth |
| Knott SRV, [78] | Breast cancer metastasis | Supports lung metastasis via EMT gene expression |
| Halbrook CJ, [79] | Pancreatic cancer | Protect tumor cells from ETC inhibition |
| Hinze L, [80] | Colorectal cancer | GSK3-dependent proteolytic scavenging to protect from L-asparaginase treatment |
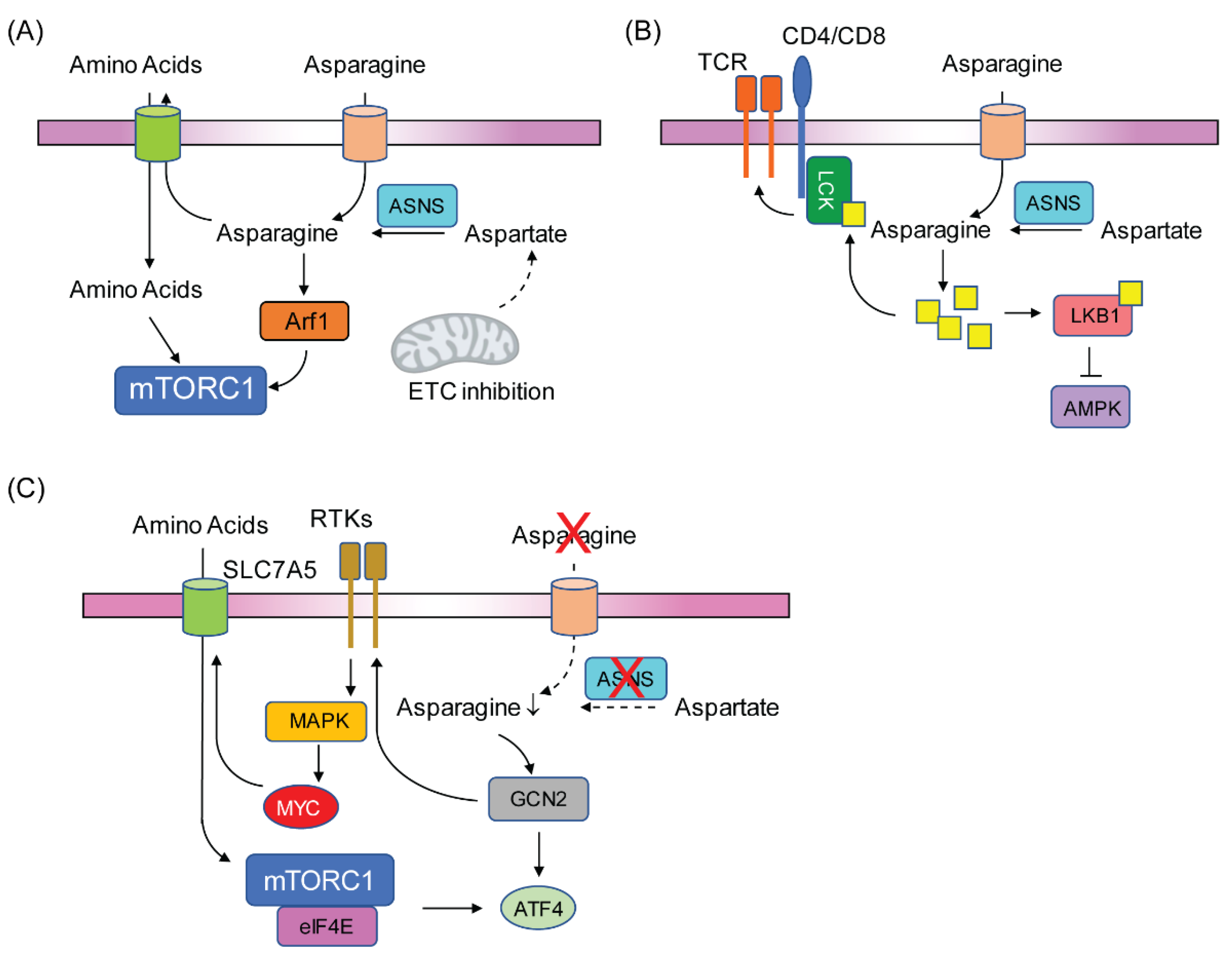
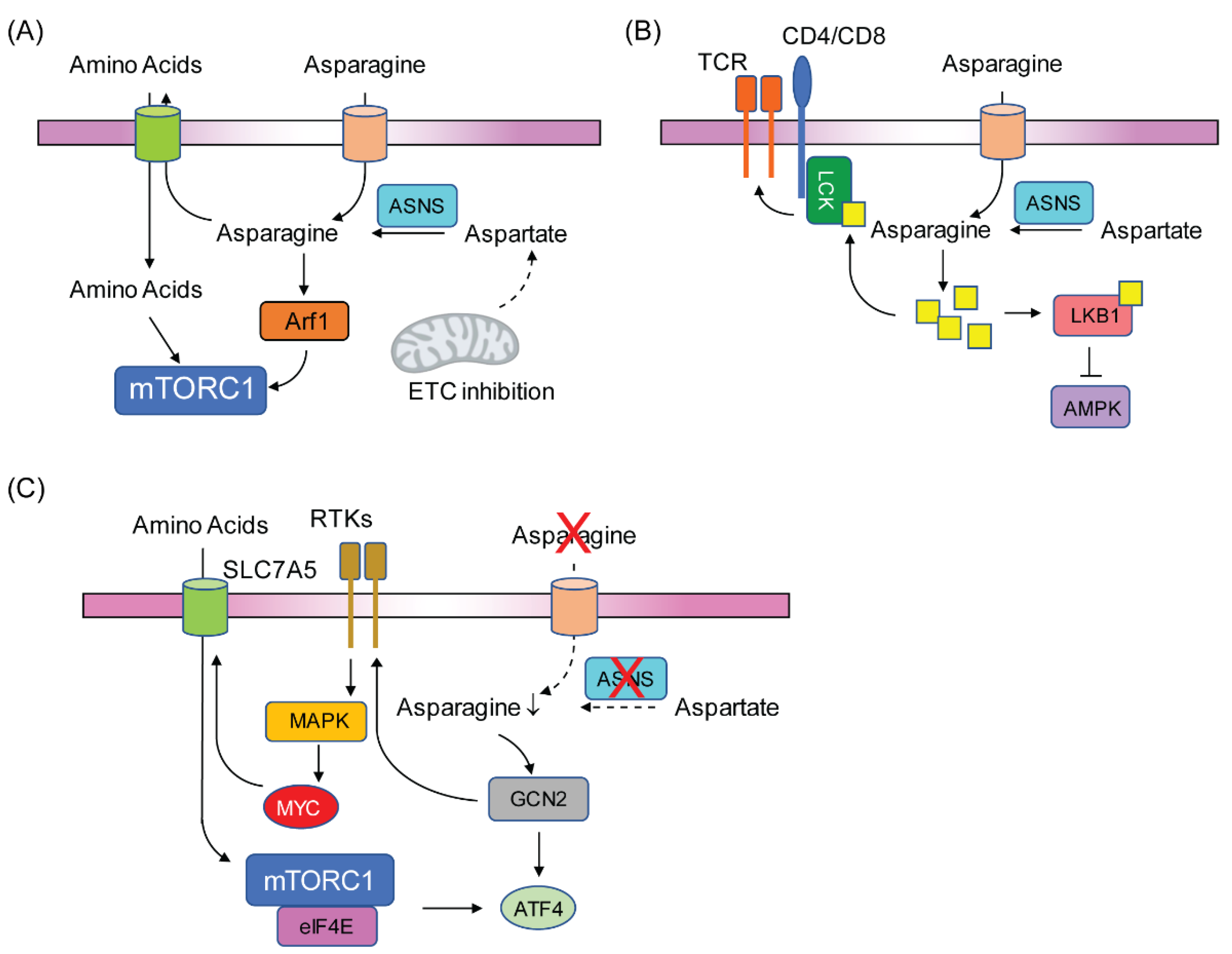
3.2. Asparagine Regulates Cancer Cell Signaling
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Palm, W.; Thompson, C.B. Nutrient acquisition strategies of mammalian cells. Nature 2017, 546, 234–242. [Google Scholar] [CrossRef]
- Choi, B.H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avramis, V.I. Asparaginases: Biochemical pharmacology and modes of drug resistance. Anticancer Res. 2012, 32, 2423–2437. [Google Scholar] [PubMed]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. I. Course of transplanted cancers of various kinds in mice and rats given guinea pig serum, horse serum, or rabbit serum. J. Exp. Med. 1953, 98, 565–582. [Google Scholar] [CrossRef]
- Kidd, J.G. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. Studies on the nature of the active serum constituent: Histological mechanism of the regression: Tests for effects of guinea pig serum on lymphoma cells in vitro: Discussion. J. Exp. Med. 1953, 98, 583–606. [Google Scholar] [PubMed]
- Broome, J.D. Evidence that the L-asparaginase activity of guinea pig serum is responsible for its antilymphoma effects. Nature 1961, 191, 2. [Google Scholar] [CrossRef]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. Lymphoma 6C3HED cells cultured in a medium devoid of L-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J. Exp. Med. 1963, 118, 121–148. [Google Scholar] [CrossRef] [Green Version]
- Broome, J.D. Evidence that the L-asparaginase of guinea pig serum is responsible for its antilymphoma effects. I. Properties of the L-asparaginase of guinea pig serum in relation to those of the antilymphoma substance. J. Exp. Med. 1963, 118, 99–120. [Google Scholar] [CrossRef] [Green Version]
- Yellin, T.O.; Wriston, J.C., Jr. Purification and properties of guinea pig serum asparaginase. Biochemistry 1966, 5, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, B.; Krakoff, I.; Burchenal, J.; Karnofsky, D.; Golbey, R.; Dowling, M.; Oettgen, H.; Lipton, A. Clinical results of treatment with E. coli L-asparaginase in adults with leukemia, lymphoma, and solid tumors. Cancer 1970, 25, 279–305. [Google Scholar] [CrossRef]
- Capizzi, R.L.; Bertino, J.R.; Skeel, R.T.; Creasey, W.A.; Zanes, R.; Olayon, C.; Peterson, R.G.; Handschumacher, R.E. L-asparaginase: Clinical, biochemical, pharmacological, and immunological studies. Ann. Intern. Med. 1971, 74, 893–901. [Google Scholar] [CrossRef]
- Abuchowski, A.; Kazo, G.M.; Verhoest, C.R., Jr.; Van Es, T.; Kefkewitz, D.; Nucci, M.L.; Viau, A.T.; Davis, F.F. Cancer therapy with chemically modified enzymes. I. Antitumor properties of polyethylene glycoL-asparaginase conjugates. Cancer Biochem. Biophys. 1984, 7, 175–186. [Google Scholar]
- Koerholz, D.; Brueck, M.; Nuenberger, W.; Juergens, H.; Goebel, U.; Wahn, V. Chemical and immunological characteristics of four different L-asparaginase preparations. Eur. J. Haematol. 1989, 42, 417–424. [Google Scholar] [CrossRef]
- Asselin, B.L.; Whitin, J.C.; Coppola, D.J.; Rupp, I.P.; Sallan, S.E.; Cohen, H.J. Comparative pharmacokinetic studies of three asparaginase preparations. J. Clin. Oncol. 1993, 11, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Ahlke, E.; Nowak-Gottl, U.; Schulze-Westhoff, P.; Werber, G.; Borste, H.; Wurthwein, G.; Jurgens, H.; Boos, J. Dose reduction of asparaginase under pharmacokinetic and pharmacodynamic control during induction therapy in children with acute lymphoblastic leukaemia. Br. J. Haematol. 1997, 96, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Vieira Pinheiro, J.P.; Ahlke, E.; Nowak-Gottl, U.; Hempel, G.; Muller, H.J.; Lumkemann, K.; Schrappe, M.; Rath, B.; Fleischhack, G.; Mann, G.; et al. Pharmacokinetic dose adjustment of Erwinia asparaginase in protocol II of the paediatric ALL/NHL-BFM treatment protocols. Br. J. Haematol. 1999, 104, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Duval, M.; Suciu, S.; Ferster, A.; Rialland, X.; Nelken, B.; Lutz, P.; Benoit, Y.; Robert, A.; Mane, A.-M.; Vilmer, E.; et al. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: Results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood 2002, 99, 2734–2739. [Google Scholar] [CrossRef] [Green Version]
- Klug Albertsen, B.; Schmiegelow, K.; Schrøder, H.; Carlsen, N.T.; Rosthøj, S.; Avramis, V.I.; Jakobsen, P. Anti-Erwinia asparaginase antibodies during treatment of childhood acute lymphoblastic leukemia and their relationship to outcome: A case-control study. Cancer Chemother. Pharm. 2002, 50, 117–120. [Google Scholar] [CrossRef]
- Boos, J.; Werber, G.; Ahlke, E.; Schultze-Westhoff, P.; Nowak-Gottl, U.; Würthwein, G.; Verspohl, E.J.; Ritter, J.; Jürgens, H. Monitoring of asparaginase activity and asparagine levels in children on different asparaginase preparations. Eur. J. Cancer 1996, 32, 1544–1550. [Google Scholar] [CrossRef]
- Angiolillo, A.L.; Schore, R.J.; Meenakshi, D.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; Keilani, T.; Lane, A.R.; Loh, M.L.; et al. Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: Results from Children’s Oncology Group Study AALL07P4. J. Clin. Oncol. 2014, 32, 3874–3882. [Google Scholar] [CrossRef] [Green Version]
- Tallal, L.; Tan, C.; Oettgen, H.; Wollner, N.; McCarthy, M.; Helson, L.; Burchenal, J.; Karnofsky, D.; Murpjy, M.L. E. coli L-asparaginase in the treatment of leukemia and solid tumors in 131 children. Cancer 1970, 25, 306–320. [Google Scholar] [CrossRef]
- Worton, K.S.; Kerbel, R.S.; Andrulis, I.L. Hypomethylation and reactivation of the asparagine synthetase gene induced by L-asparaginase and ethyl methanesulfonate. Cancer Res. 1991, 51, 985–989. [Google Scholar]
- Ren, Y.; Soy, S.; Ding, Y.; Iqbal, J.; Broome, J.D. Methylation of the asparagine synthetase promoter in human leukemic cell lines is associated with a specific methyl binding protein. Oncogene 2004, 23, 3953–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigouin, C.; Nguyen, H.A.; Schalk, A.M.; Lavie, A. Discovery of human-like L-asparaginases with potential clinical use by directed evolution. Sci. Rep. 2017, 7, 10224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avramis, V.I.; Sencer, S.; Periclou, A.P.; Sather, H.; Bostrom, B.C.; Cohen, L.J.; Ettinger, A.G.; Ettinger, L.J.; Franklin, J.; Gaynon, P.S.; et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: A Children’s Cancer Group study. Blood 2002, 99, 1986–1994. [Google Scholar] [CrossRef] [Green Version]
- Jarrar, M.; Sencer, S.; Periclou, A.P.; Sather, H.; Bostrom, B.C.; Cohen, L.J.; Ettinger, A.G.; Ettinger, L.J.; Franklin, J.; Gaynon, P.S.; et al. Asparagine depletion after pegylated E. coli asparaginase treatment and induction outcome in children with acute lymphoblastic leukemia in first bone marrow relapse: A Children’s Oncology Group study (CCG-1941). Pediatr. Blood Cancer 2006, 47, 141–146. [Google Scholar] [CrossRef]
- Silverman, L.B.; Supko, J.G.; Stevenson, K.; Woodward, C.; Vrooman, V.M.; Neuberg, D.S.; Asselin, B.L.; Athale, U.H.; Clavell, L.; Cole, P.D.; et al. Intravenous PEG-asparaginase during remission induction in children and adolescents with newly diagnosed acute lymphoblastic leukemia. Blood 2010, 115, 1351–1353. [Google Scholar] [CrossRef] [Green Version]
- Douer, D.; Yampolsky, H.; Cohen, L.J.; Watkins, K.; Levine, A.M.; Periclou, A.P.; Avramis, V.I. Pharmacodynamics and safety of intravenous pegaspargase during remission induction in adults aged 55 years or younger with newly diagnosed acute lymphoblastic leukemia. Blood 2007, 109, 2744–2750. [Google Scholar] [CrossRef] [Green Version]
- Holcenberg, J.S.; Teller, D.C. Physical properties of antitumor glutaminase-asparaginase from Pseudomonas 7A. J. Biol. Chem. 1976, 251, 5375–5380. [Google Scholar] [CrossRef]
- Panosyan, E.H.; Grigoryan, R.S.; Avramis, I.A.; Seibel, N.L.; Gaynon, P.S.; Siegel, S.E.; Fingert, H.J.; Avramis, V.I. Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer Res. 2004, 24, 1121–1125. [Google Scholar]
- Nguyen, H.A.; Su, Y.; Zhang, J.Y.; Antanasijevic, A.; Caffrey, M.; Schalk, A.M.; Liu, L.; Rondelli, R.; Oh, A.; Mahmud, D.L.; et al. A Novel L-Asparaginase with low L-Glutaminase Coactivity Is Highly Efficacious against Both T- and B-cell Acute Lymphoblastic Leukemias In vivo. Cancer Res. 2018, 78, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.K.; Horvath, T.D.; Tan, L.; Link, T.; Harutyunyan, K.G.; Pontikos, M.A.; Anishkin, A.; Du, D.; Martin, L.A.; Yin, E.; et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity against Acute Lymphoblastic Leukemia. Mol. Cancer Ther. 2019, 18, 1587–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.K.; Lorenzi, P.L.; Anishkin, A.; Purwaha, P.; Rogers, D.M.; Sukharev, S.; Rempe, S.B.; Weinstein, J.N. The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 2014, 123, 3596–3606. [Google Scholar] [CrossRef] [Green Version]
- Silverman, L.B.; Declerck, L.; Gelber, R.D.; Kimball, D.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Huwitz, C.A.; Moghrabi, A.; Samson, Y.; et al. Results of Dana-Farber Cancer Institute Consortium protocols for children with newly diagnosed acute lymphoblastic leukemia (1981–1995). Leukemia 2000, 14, 2247–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abshire, T.C.; Pollock, B.H.; Billett, A.L.; Bradley, P.; Buchanan, G.R. Weekly polyethylene glycol conjugated L-asparaginase compared with biweekly dosing produces superior induction remission rates in childhood relapsed acute lymphoblastic leukemia: A Pediatric Oncology Group Study. Blood 2000, 96, 1709–1715. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Pollock, B.H.; Billett, A.L.; Bradley, P.; Buchanan, G.R. Asparaginase pharmacokinetics after intensive polyethylene glycoL-conjugated L-asparaginase therapy for children with relapsed acute lymphoblastic leukemia. Clin. Cancer Res. 2004, 96, 5335–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, S.S.; Holdsworth, M.T.; Devidas, M.; Reisch, D.W.; Chauvenet, A.; Ravindranath, A.; Ducore, J.M.; Amyhlon, M.D. Antimetabolite-based therapy in childhood T-cell acute lymphoblastic leukemia: A report of POG study 9296. Pediatr. Blood Cancer 2006, 46, 179–186. [Google Scholar] [CrossRef]
- Yang, L.; Panetta, J.C.; Cai, X.; Yang, W.; Pei, D.; Cheng, C.; Konegay, N.; Pui, C.-H.; Relling, M.V. Asparaginase may influence dexamethasone pharmacokinetics in acute lymphoblastic leukemia. J. Clin. Oncol. 2008, 26, 1932–1939. [Google Scholar] [CrossRef]
- Schrappe, M.; Reiter, A.; Ludwig, W.D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000, 95, 3310–3322. [Google Scholar]
- Amylon, M.D.; Shuster, J.; Pullen, J.; Berard, C.; Link, M.P.; Wharam, M.; Katz, J.; Yu, A.; Laver, J.; Ravindranath, Y.; et al. Intensive high-dose asparaginase consolidation improves survival for pediatric patients with T cell acute lymphoblastic leukemia and advanced stage lymphoblastic lymphoma: A Pediatric Oncology Group study. Leukemia 1999, 13, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzer, W.L.; Devidas, M.; Carroll, W.L.; Winick, N.; Pullen, J.; Hunger, S.P.; Camitta, B.A. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984–2001: A report from the children’s oncology group. Leukemia 2010, 24, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, W.E.; Tsiatis, A.; Rivera, G.; Murphy, S.B.; Dahl, G.V.; Denison, M.; Crom, V.R.; Barker, L.F.; Mauer, A.M. Anaphylactoid reactions to Escherichia coli and Erwinia asparaginase in children with leukemia and lymphoma. Cancer 1982, 49, 1378–1383. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.K.; Chau, I.Y.; Coccia, P.F. Antibody response to Escherichia coli L-asparaginase. Prognostic significance and clinical utility of antibody measurement. Am. J. Pediatr. Hematol. Oncol. 1986, 8, 99–104. [Google Scholar]
- Panosyan, E.H.; Seibel, N.L.; Martin-Aragon, S.; Gaynon, P.S.; Avramis, I.A.; Sather, H.; Franklin, J.; Nachman, J.; Ettinger, L.; Mei, L.A.; et al. Asparaginase antibody and asparaginase activity in children with higher-risk acute lymphoblastic leukemia: Children’s Cancer Group Study CCG-1961. J. Pediatr. Hematol. Oncol. 2004, 26, 217–226. [Google Scholar] [CrossRef]
- van der Meer, L.T.; Terry, S.Y.; van Ingen Schenau, D.S.; Andree, K.C.; Franssen, G.M.; Roeleveld, D.M.; Metselaar, J.M.; Reinheckel, T.; Hoogerbrugge, P.M.; Boerman, O.C.; et al. In vivo Imaging of Antileukemic Drug Asparaginase Reveals a Rapid Macrophage-Mediated Clearance from the Bone Marrow. J. Nucl. Med. 2017, 58, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, M.N.; Butterworth, E.A.; Kilberg, M.S. Asparagine synthetase: Regulation by cell stress and involvement in tumor biology. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E789–E799. [Google Scholar] [CrossRef] [Green Version]
- Aslanian, A.M.; Fletcher, B.S.; Kilberg, M.S. Asparagine synthetase expression alone is sufficient to induce L-asparaginase resistance in MOLT-4 human leukaemia cells. Biochem. J. 2001, 357, 321–328. [Google Scholar] [CrossRef]
- Ding, Y.; Li, Z.; Broome, J.D. Epigenetic changes in the repression and induction of asparagine synthetase in human leukemic cell lines. Leukemia 2005, 19, 420–426. [Google Scholar] [CrossRef]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Srivastava, S.; Seim, G.; Pavlova, N.N.; King, B.; Zou, L.; Zhang, C.; Zhong, M.; Feng, H.; Kapur, R.; et al. Promoter demethylation of the asparagine synthetase gene is required for ATF4-dependent adaptation to asparagine depletion. J. Biol. Chem. 2019, 294, 18674–18684. [Google Scholar] [CrossRef]
- Chen, H.; Pan, Y.X.; Dudenhausen, E.E.; Kilberg, M.S. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J. Biol. Chem. 2004, 279, 50829–50839. [Google Scholar]
- Williams, R.T.; Guarecuco, R.; Gates, L.A.; Barrows, D.; Passarelli, M.C.; Carey, B.; Baudrier, L.; Jeewajee, S.; La, K.; Prozer, B.; et al. ZBTB1 Regulates Asparagine Synthesis and Leukemia Cell Response to L-Asparaginase. Cell Metab. 2020, 31, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Aslanian, A.M.; Kilberg, M.S. Multiple adaptive mechanisms affect asparagine synthetase substrate availability in asparaginase-resistant MOLT-4 human leukaemia cells. Biochem. J. 2001, 358, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nagel, R.; Zaal, E.A.; Ugalde, A.P.; Han, R.; Proost, N.; Song, J.-Y.; Pataskar, A.; Burylo, A.; Fu, H.; et al. SLC1A3 contributes to L-asparaginase resistance in solid tumors. EMBO J. 2019, 38, e102147. [Google Scholar] [CrossRef]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stams, W.A.; den Boer, M.L.; Beverloo, H.B.; Meijerink, J.P.; Stigter, R.L.; van Wering, E.R.; Janka-Schaub, G.E.; Slater, R.; Pieters, R. Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 2003, 101, 2743–2747. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Pan, Y.X.; Zhou, M.; Harvey, R.C.; Hunger, S.P.; Kilberg, M.S. Correlation between asparaginase sensitivity and asparagine synthetase protein content, but not mRNA, in acute lymphoblastic leukemia cell lines. Pediatr. Blood Cancer 2008, 50, 274–279. [Google Scholar] [CrossRef]
- Touzart, A.; Lengliné, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic silencing affects L-asparaginase sensitivity and predicts outcome in T-ALL. Clin. Cancer Res. 2019, 25, 2483–2493. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef]
- Ehsanipour, E.A.; Sheng, X.; Behan, J.W.; Wang, X.; Butturini, A.; Avramis, V.I.; Mittelman, S.D. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013, 73, 2998–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinze, L.; Pfirrmann, M.; Karim, S.; Degar, J.; McGuckin, C.; Vinjamur, D.; Sacher, J.; Stevenson, K.E.; Neuberg, D.S.; Orellana, E.; et al. Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias. Cancer Cell 2019, 35, 664–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes. Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Inoue, J.; Sakakguchi, K.; Takagi, M.; Mizutani, S.; Inazawa, J. Autophagy is required for cell survival under L-asparaginase-induced metabolic stress in acute lymphoblastic leukemia cells. Oncogene 2017, 36, 4267–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kang, S.; Wang, X.; Rosales, J.L.; Gao, X.; Byun, H.G.; Jin, Y.; Fu, S.; Wang, J.; Lee, K.-Y. HAP1 loss confers L-asparaginase resistance in ALL by downregulating the calpain-1-Bid-caspase-3/12 pathway. Blood 2019, 133, 2222–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holleman, A.; Cheok, M.H.; den Boer, M.L.; Yang, W.; Veerman, A.J.; Kazemier, K.M.; Pei, D.; Cheng, C.; Pui, C.-H.; Relling, M.V.; et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med. 2004, 351, 533–542. [Google Scholar] [CrossRef]
- Holleman, A.; den Boer, M.L.; de Menezes, R.X.; Cheok, M.H.; Cheng, C.; Kazemier, K.M.; Janka-Schaub, G.E.; Gobel, U.; Graubner, U.B.; Evans, V.E.; et al. The expression of 70 apoptosis genes in relation to lineage, genetic subtype, cellular drug resistance, and outcome in childhood acute lymphoblastic leukemia. Blood 2006, 107, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Holleman, A.; Boer, M.L.D.; Kazemier, K.M.; Beverloo, H.B.; von Bergh, A.R.; Janka-Schaub, G.E.; Pieters, R. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood 2005, 106, 1817–1823. [Google Scholar] [CrossRef]
- Rousseau, J.; Gagné, V.; Labuda, M.; Beaubois, C.; Sinnett, D.; Laverdière, C.; Moghrabi, A.; Sallan, S.E.; Silverman, L.B.; Neuberg, D.; et al. ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood 2011, 118, 5883–5890. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Rosales, J.L.; Meier-Stephenson, V.; Kim, S.; Lee, K.Y.; Narendran, A. Genome-wide loss-of-function genetic screening identifies opioid receptor mu1 as a key regulator of L-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene 2017, 36, 5910–5913. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Brüning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, T.; Ramos da Silva, S.; Lee, J.J.; Lu, C.; Eoh, H.; Jung, J.U.; Gao, S.-J. A Critical Role of Glutamine and Asparagine gamma-Nitrogen in Nucleotide Biosynthesis in Cancer Cells Hijacked by an Oncogenic Virus. MBio 2017, 8, e01179–e01217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107. [Google Scholar] [CrossRef] [Green Version]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.-K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017, 26, 817–829. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.-O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Halbrook, C.J.T.G.; McCarthy, A.; Nelson, B.S.; Sajjakulnukit, P.; Krall, A.S.; Mullen, P.J.; Zhang, L.; Batra, S.; Viale, A.; Stanger, B.Z.; et al. Clonal Heterogeneity Supports Mitochondrial Metabolism in Pancreatic Cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hinze, L.; Labrosse, R.; Degar, J.; Han, T.; Schatoff, E.M.; Schreek, S.; Karim, S.; McGuckin, C.; Sacher, J.R.; Wagner, F.; et al. Exploiting the Therapeutic Interaction of WNT Pathway Activation and Asparaginase for Colorectal Cancer Therapy. Cancer Discov. 2020, 10, 1690–1705. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krall, A.S.; Mullen, P.J.; Surjono, F.; Momcilovic, M.; Schmid, E.W.; Halbrook, C.J.; Thambundit, A.; Mittelman, S.D.; Lyssiotis, C.A.; Shackelford, D.B.; et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021, 33, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Yao, P.; Li, L.; Ji, F.; Zhao, S.; Xu, C.; Lan, X.; Jiang, P. p53-mediated control of aspartate-asparagine homeostasis dictates LKB1 activity and modulates cell survival. Nat. Commun. 2020, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, G.; Li, L.; Li, D.; Dong, Z.; Jiang, P. Asparagine enhances LCK signalling to potentiate CD8(+) T-cell activation and anti-tumour responses. Nat. Cell Biol. 2021, 23, 75–86. [Google Scholar] [CrossRef]
- Pathria, G.; Lee, J.S.; Hasnis, E.; Tandoc, K.; Scott, D.A.; Verma, S.; Feng, Y.; Larue, L.; Sahu, A.D.; Topisirovic, I. Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat. Cell Biol. 2019, 21, 1590–1603. [Google Scholar] [CrossRef]
- Pathria, G.; Verma, S.; Yin, J.; Scott, D.A.; Ronai, Z.E.A. MAPK signaling regulates c-MYC for melanoma cell adaptation to asparagine restriction. EMBO Rep. 2021, 22, e51436. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, J.; Batra, S.; Zhang, J. Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites 2021, 11, 402. https://doi.org/10.3390/metabo11060402
Jiang J, Batra S, Zhang J. Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites. 2021; 11(6):402. https://doi.org/10.3390/metabo11060402
Chicago/Turabian StyleJiang, Jie, Sandeep Batra, and Ji Zhang. 2021. "Asparagine: A Metabolite to Be Targeted in Cancers" Metabolites 11, no. 6: 402. https://doi.org/10.3390/metabo11060402
APA StyleJiang, J., Batra, S., & Zhang, J. (2021). Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites, 11(6), 402. https://doi.org/10.3390/metabo11060402