
Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
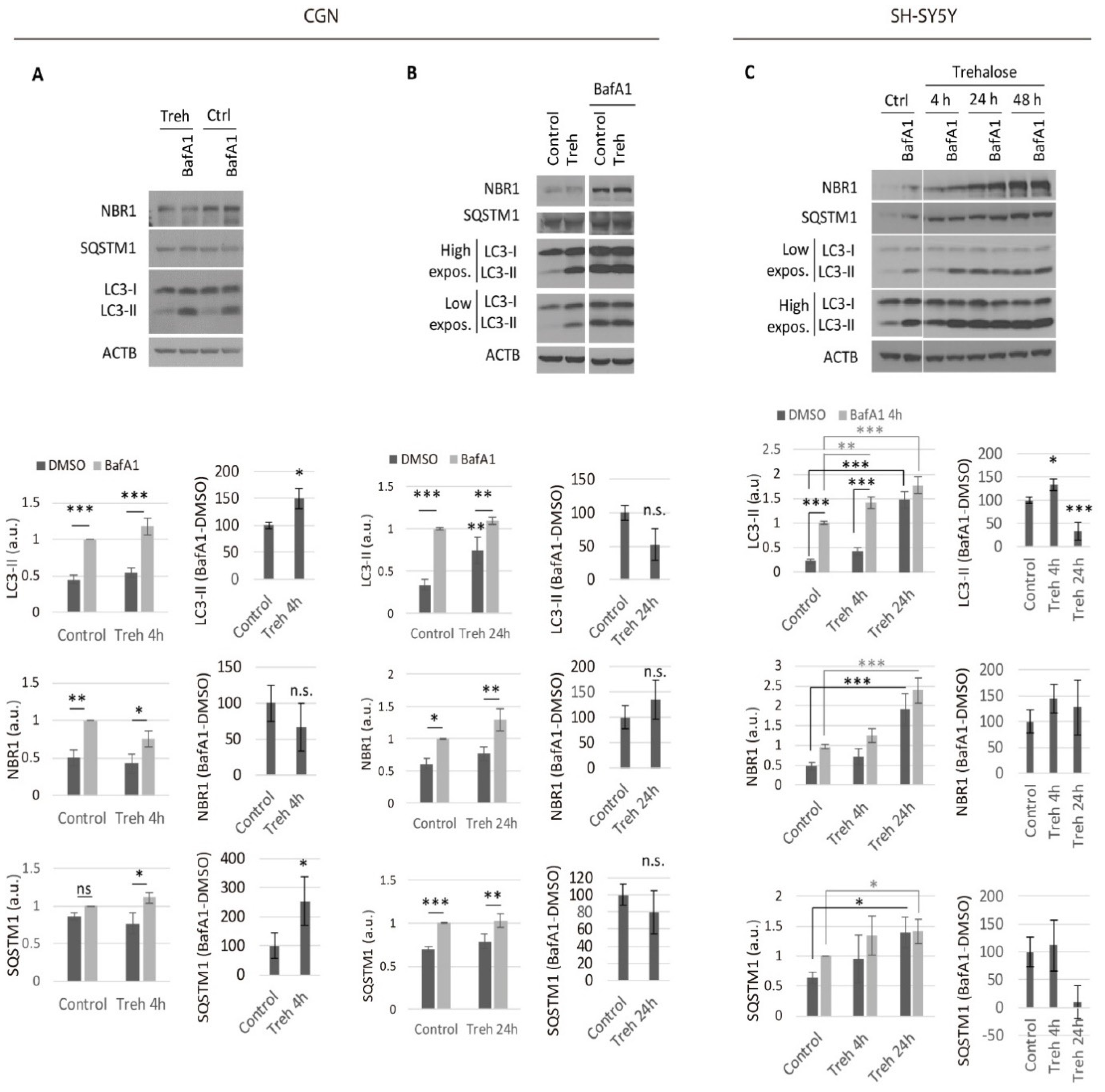
2.1. Trehalose Only Increases the Neuronal Autophagic Flux in the Short Term
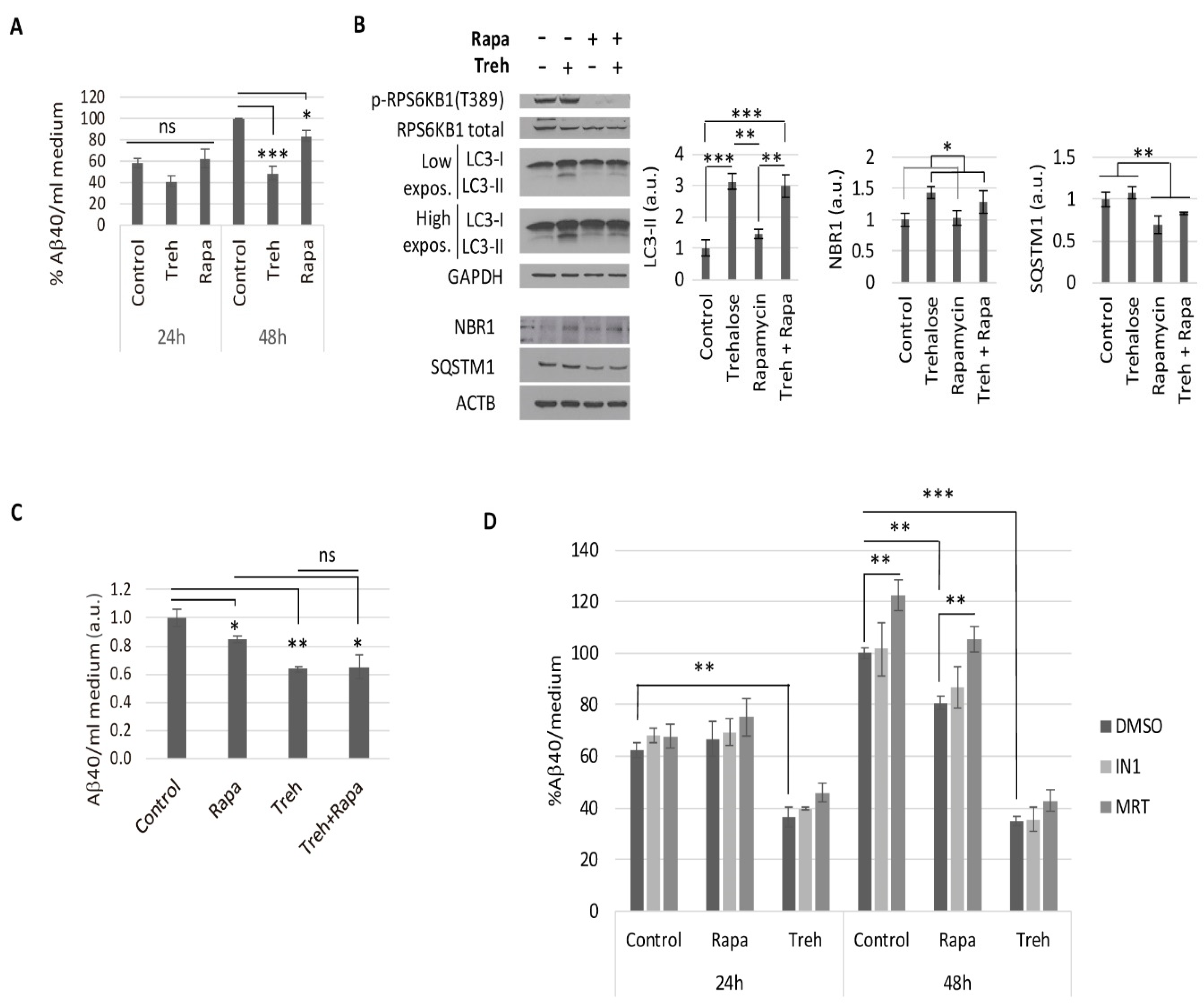
2.2. Trehalose Reduces Aβ40 Secretion in Primary Neurons Isolated from APP/PSEN1 Mice in an Autophagy Independent Fashion
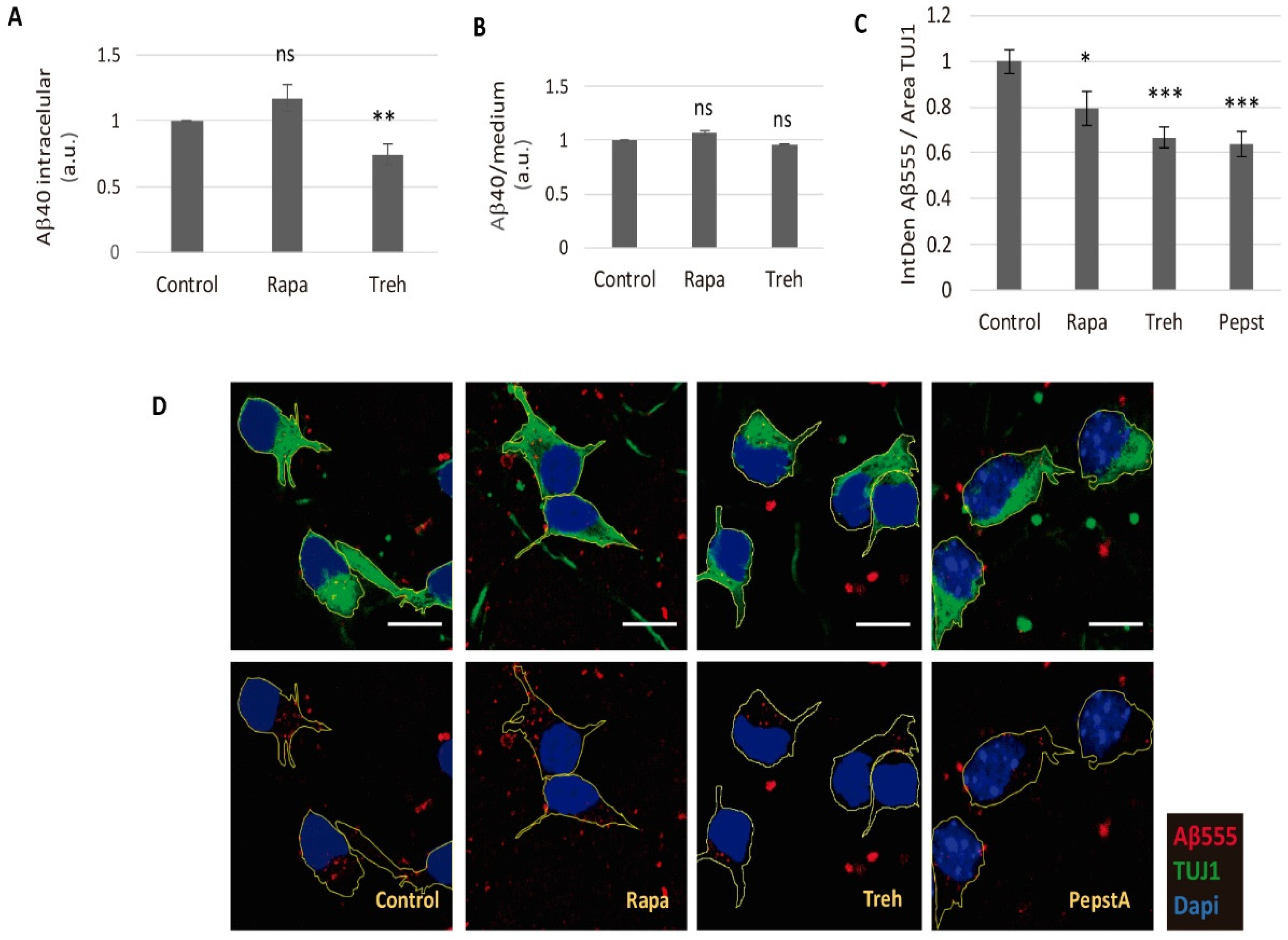
2.3. Lowered Secreted Levels of Aβ40 by Trehalose Are Not Due to Its Intracellular Storage or to a Higher Endocytosis Rate
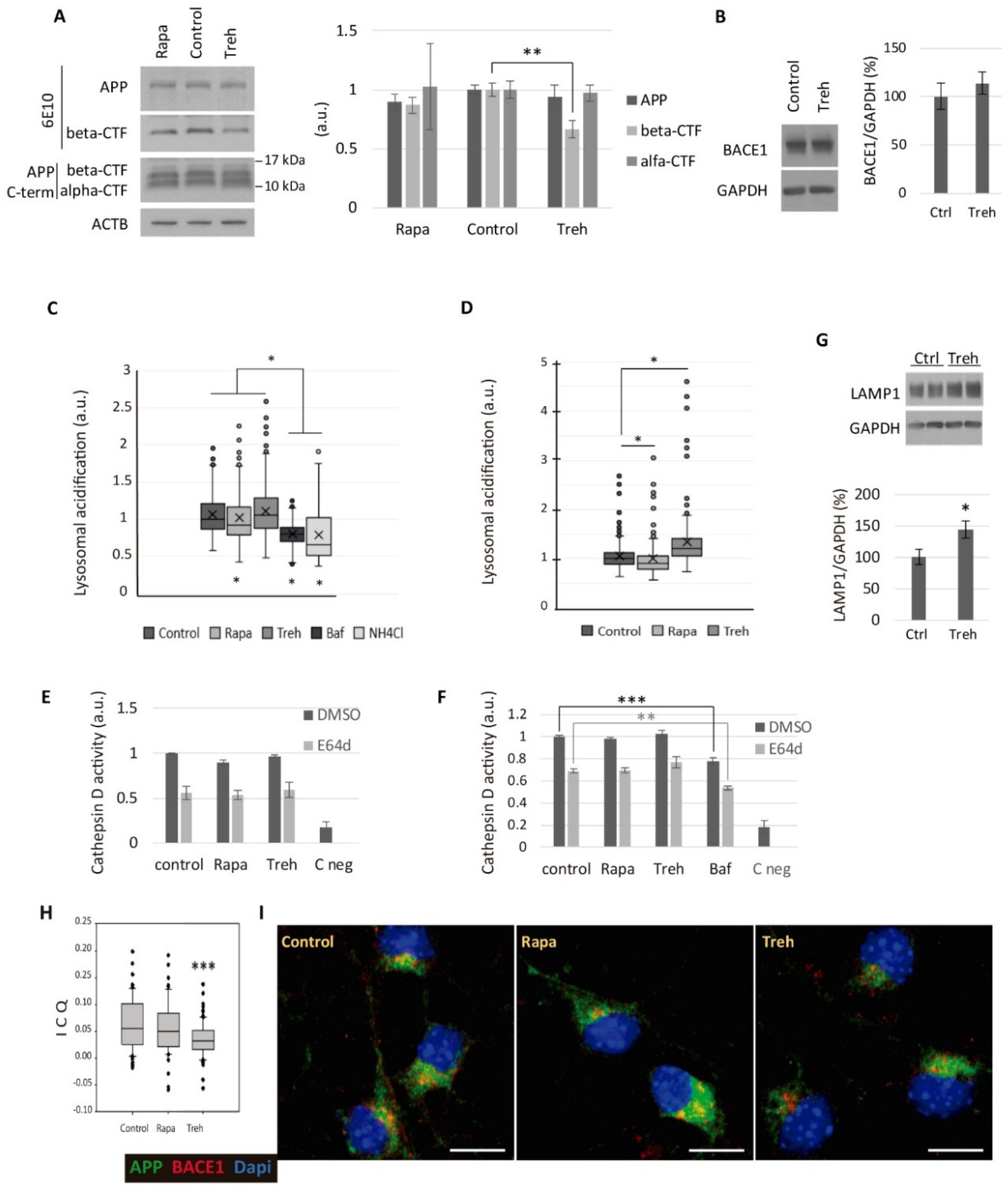
2.4. Trehalose Diminishes the Amyloidogenic Processing of APP
3. Discussion
4. Materials and Methods
4.1. Animal Handling
4.2. Cell Culture
4.3. Autophagic Flux
4.4. Gel Electrophoresis and Western Blots
4.5. Aβ Quantification by ELISA
4.6. Immunofluorescence
4.7. Aβ Endocytosis Assay
4.8. LysoSensor Assay
4.9. Cathepsin D Activity Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sarkar, S.; Rubinsztein, D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst. 2008, 4, 895–901. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 23, 5641–5652. [Google Scholar] [CrossRef]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Ertmer, A.; Schätzl, H.M. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef]
- Yang, C.R.; Yu, R.K. Intracerebral transplantation of neural stem cells combined with trehalose ingestion alleviates pathology in a mouse model of Huntington’s disease. J. Neurosci. Res. 2009, 87, 26–33. [Google Scholar] [CrossRef]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain A J. Neurol. 2012, 135 Pt 7, 2169–2177. [Google Scholar] [CrossRef]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef]
- Mayer, A.L.; Higgins, C.B.; Heitmeier, M.R.; Kraft, T.E.; Qian, X.; Crowley, J.R.; Hyrc, K.L.; Beatty, W.L.; Yarasheski, K.E.; Hruz, P.W.; et al. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci. Rep. 2016, 6, 38586. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Khalifeh, M.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Trehalose against Alzheimer’s Disease: Insights into a Potential Therapy. BioEssays News Rev. Mol. Cell. Dev. Biol. 2020, 42, e1900195. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Park, C.B.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Ariosa, A.R.; Klionsky, D.J. Autophagy core machinery: Overcoming spatial barriers in neurons. J. Mol. Med. (Berl. Ger.) 2016, 94, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 37, 377–388. [Google Scholar] [CrossRef]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648 Pt B, 571–579. [Google Scholar] [CrossRef]
- Tagliaferro, P.; Burke, R.E. Retrograde Axonal Degeneration in Parkinson Disease. J. Parkinsons Dis. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Araki, W. Post-translational regulation of the beta-secretase BACE1. Brain Res. Bull. 2016, 126 Pt 2, 170–177. [Google Scholar] [CrossRef]
- Benito-Cuesta, I.; Ordonez-Gutierrez, L.; Wandosell, F. AMPK activation does not enhance autophagy in neurons in contrast to MTORC1 inhibition: Different impact on beta-amyloid clearance. Autophagy 2020, 17, 1–16. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117 Pt 13, 2805–2812. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Bago, R.; Malik, N.; Munson, M.J.; Prescott, A.R.; Davies, P.; Sommer, E.; Shpiro, N.; Ward, R.; Cross, D.; Ganley, I.G.; et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 2014, 463, 413–427. [Google Scholar] [CrossRef]
- Bilanges, B.; Vanhaesebroeck, B. Cinderella finds her shoe: The first Vps34 inhibitor uncovers a new PI3K-AGC protein kinase connection. Biochem. J. 2014, 464, e7–e10. [Google Scholar] [CrossRef]
- Petherick, K.J.; Conway, O.J.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 28726. [Google Scholar] [CrossRef]
- Athanassakis, I.; Protopapadakis, E.; Vassiliadis, S. Localization of pepstatin’s inhibitory action during Fc-mediated antibody internalization: Possible implications for antibody-mediated viral transmission. Cell. Immunol. 2000, 199, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Ziv, E. Kinetic properties of cathepsin D and BACE 1 indicate the need to search for additional beta-secretase candidate(s). Biol. Chem. 2008, 389, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Matha, V.; Derocq, D.; Prebois, C.; Katunuma, N.; Liaudet-Coopman, E. Processing of human cathepsin D is independent of its catalytic function auto-activation: Involvement of cathepsins, L and B. J. Biochem. 2006, 139, 363–371. [Google Scholar] [CrossRef]
- Luyckx, J.; Baudouin, C. Trehalose: An intriguing disaccharide with potential for medical application in ophthalmology. Clin. Ophthalmol. 2011, 5, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.T.; Karaca, I.; Tamboli, I.Y.; Walter, J. Trehalose Alters Subcellular Trafficking and the Metabolism of the Alzheimer-associated Amyloid Precursor Protein. J. Biol. Chem. 2016, 291, 10528–10540. [Google Scholar] [CrossRef]
- Das, U.; Scott, D.A.; Ganguly, A.; Koo, E.H.; Tang, Y.; Roy, S. Activity-Induced Convergence of APP and BACE-1 in Acidic Microdomains via an Endocytosis-Dependent Pathway. Neuron 2013, 79, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Wang, L.; Ganguly, A. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Wada, S.-I.; Sawa, R.; Ohba, S.-I.; Hayashi, C.; Umekita, M.; Shibuya, Y.; Iijima, K.; Iwanami, F.; Igarashi, M. Stability and Bioavailability of Lentztrehaloses A, B, and C as Replacements for Trehalose. J. Agric. Food Chem. 2016, 64, 7121–7126. [Google Scholar] [CrossRef]
- Ordonez-Gutierrez, L.; Anton, M.; Wandosell, F. Peripheral amyloid levels present gender differences associated with aging in AbetaPP/PS1 mice. J. Alzheimer’s Dis. JAD 2015, 44, 1063–1068. [Google Scholar] [CrossRef]
- Ordonez-Gutierrez, L.; Fernandez-Perez, I.; Herrera, J.L.; Anton, M.; Benito-Cuesta, I.; Wandosell, F. Abeta PP/PS1 Transgenic Mice Show Sex Differences in the Cerebellum Associated with Aging. J. Alzheimer’s Dis. JAD 2016, 54, 645–656. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Mullins, S.R.; Sameni, M.; Blum, G.; Bogyo, M.; Sloane, B.F.; Moin, K. Three-dimensional cultures modeling premalignant progression of human breast epithelial cells: Role of cysteine cathepsins. Biol. Chem. 2012, 393, 1405–1416. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benito-Cuesta, I.; Ordoñez-Gutierrez, L.; Wandosell, F. Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction. Metabolites 2021, 11, 421. https://doi.org/10.3390/metabo11070421
Benito-Cuesta I, Ordoñez-Gutierrez L, Wandosell F. Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction. Metabolites. 2021; 11(7):421. https://doi.org/10.3390/metabo11070421
Chicago/Turabian StyleBenito-Cuesta, Irene, Lara Ordoñez-Gutierrez, and Francisco Wandosell. 2021. "Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction" Metabolites 11, no. 7: 421. https://doi.org/10.3390/metabo11070421
APA StyleBenito-Cuesta, I., Ordoñez-Gutierrez, L., & Wandosell, F. (2021). Trehalose Reduces the Secreted Beta-Amyloid Levels in Primary Neurons Independently of Autophagy Induction. Metabolites, 11(7), 421. https://doi.org/10.3390/metabo11070421