
Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases



Abstract
:
1. Introduction
2. Results
2.1. Overview of KEGG and MetaCyc Databases
2.2. Results of Compound Harmonization across KEGG and MetaCyc Databases
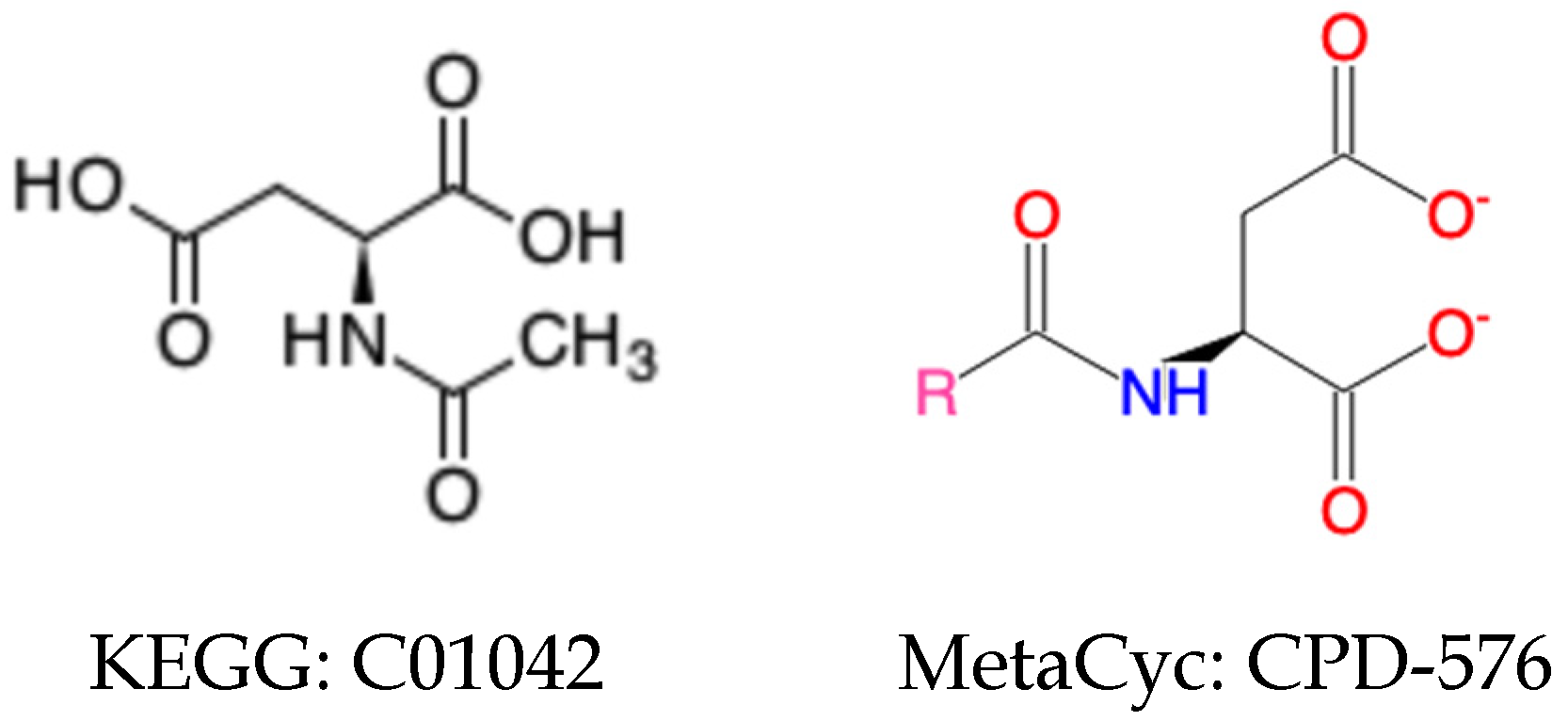
2.2.1. Harmonization of Specific Compounds
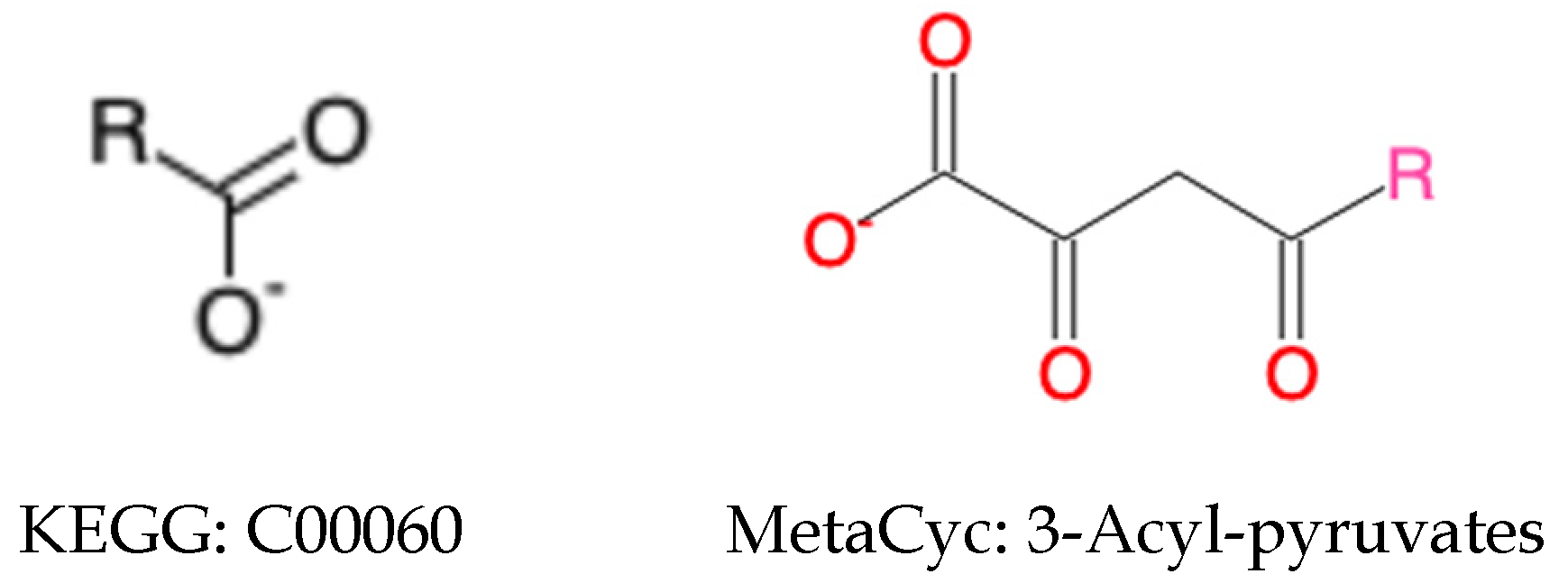
2.2.2. Harmonization of Generic Compounds
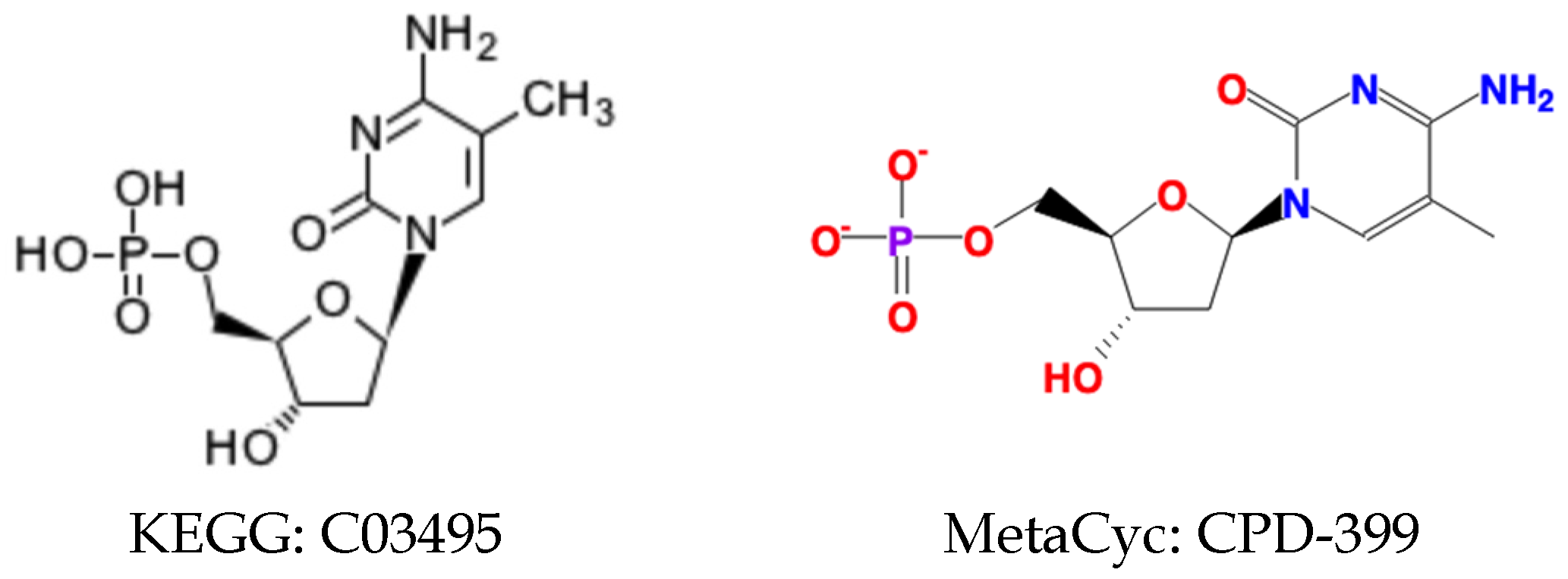
2.2.3. Harmonization of Compounds with Changeable Representations
2.2.4. Summary of Compound Harmonization
2.3. Results of Reaction Harmonization across KEGG and MetaCyc Databases
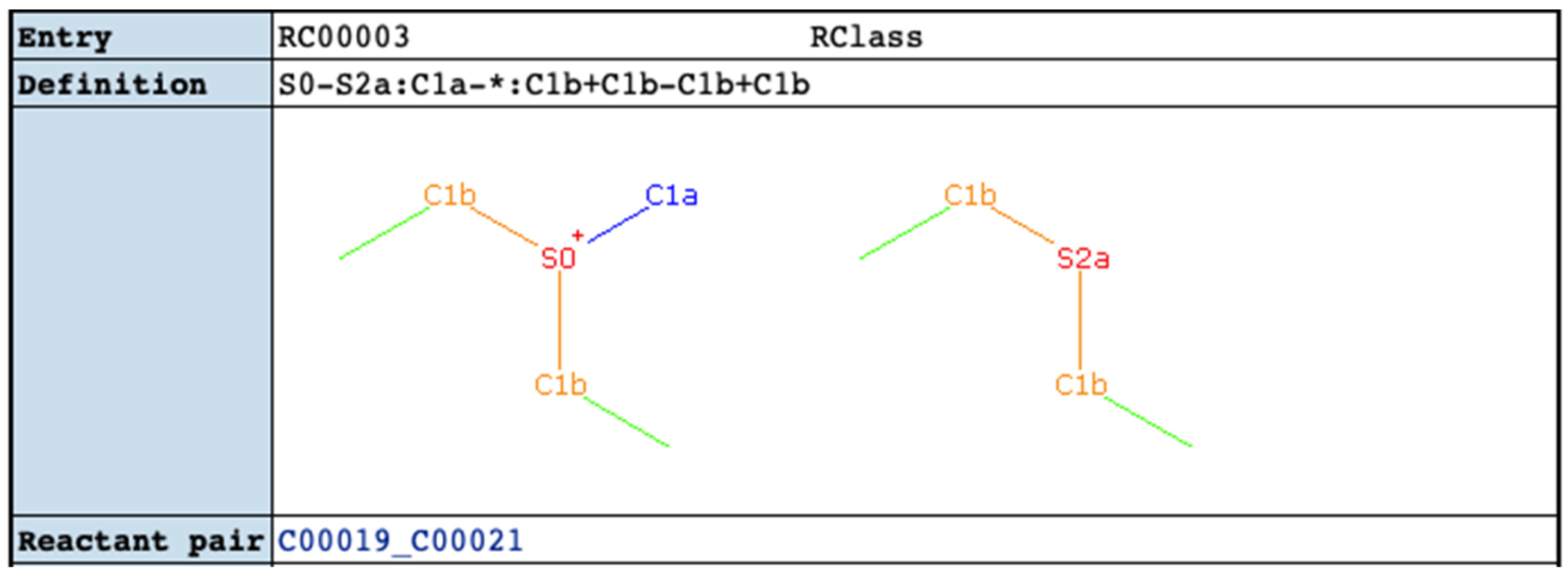
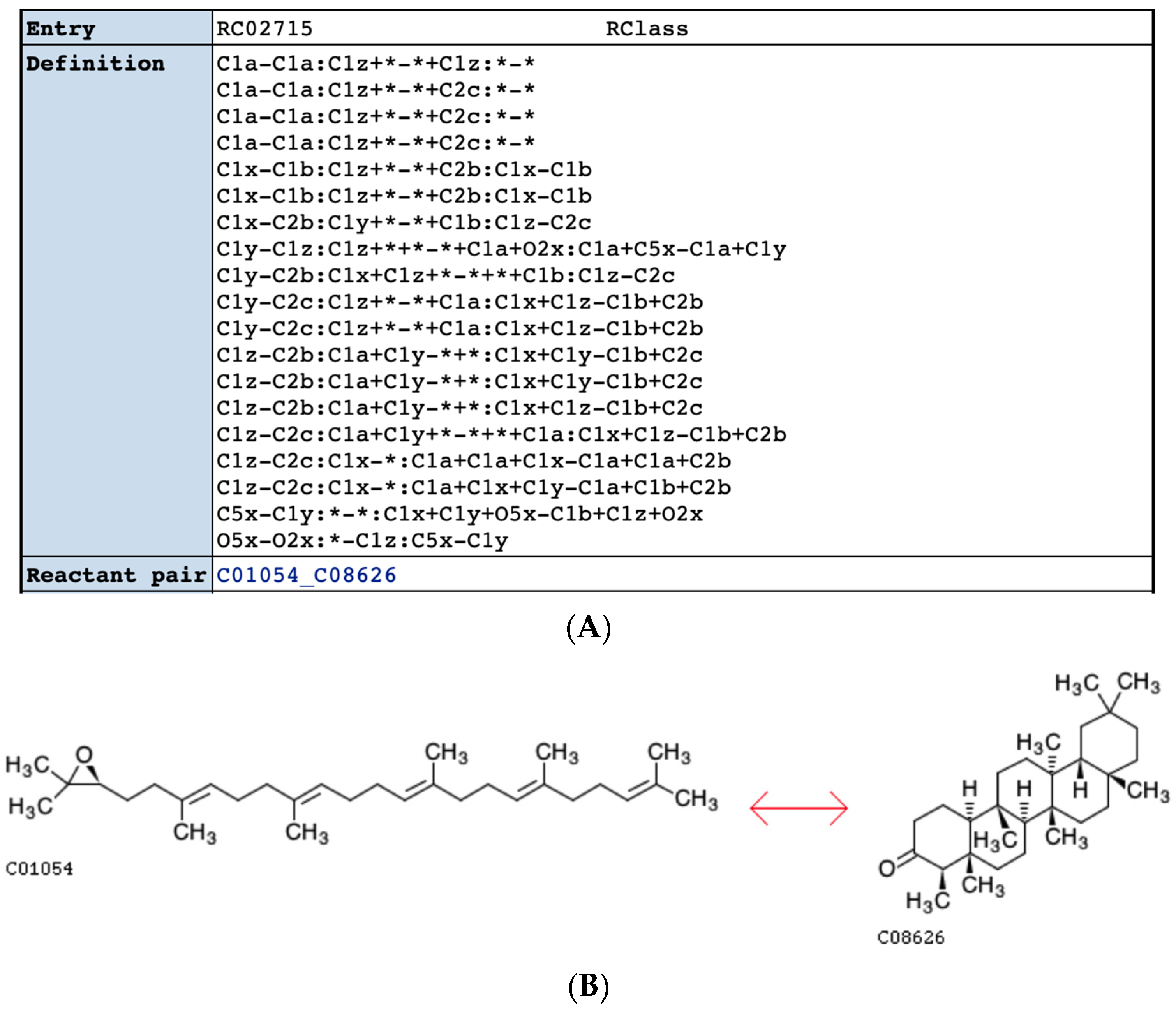
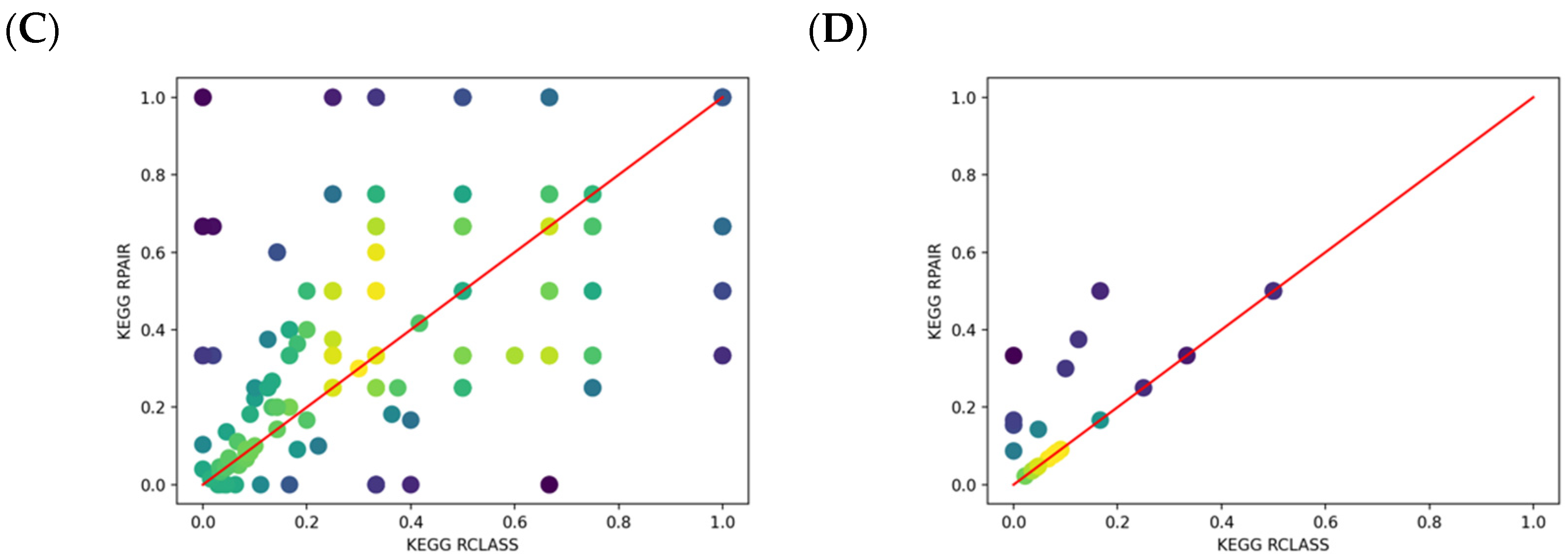
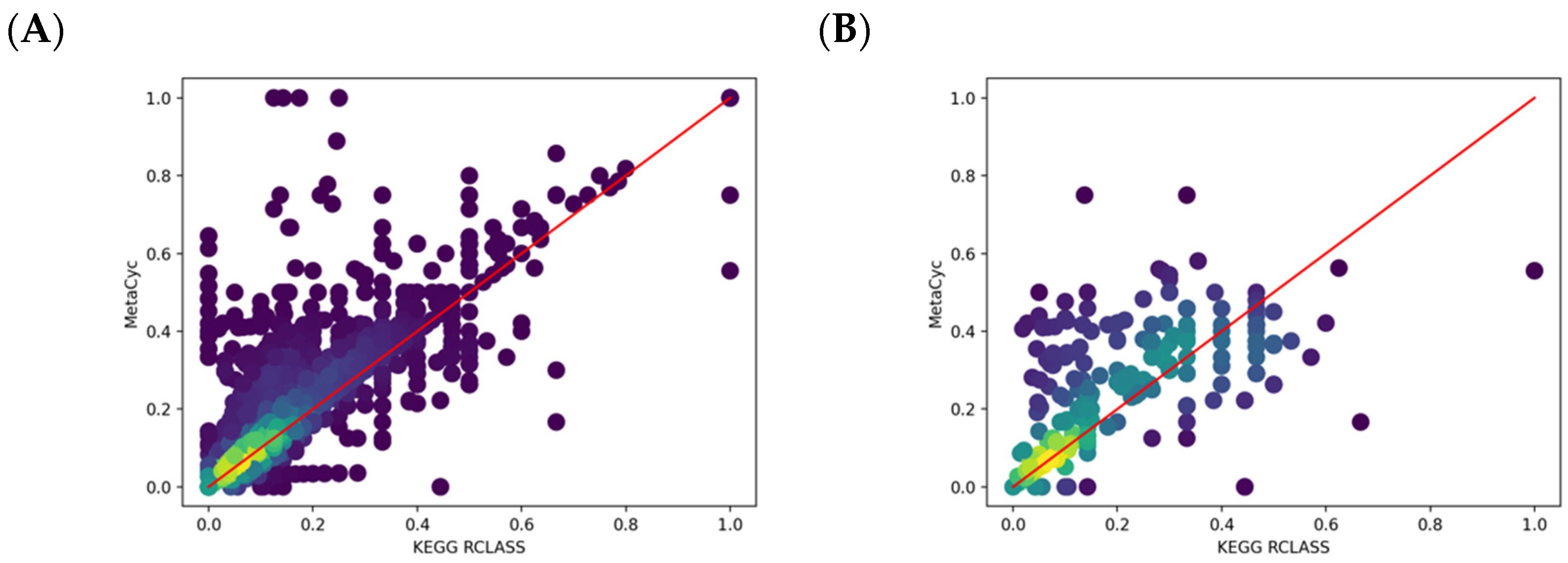
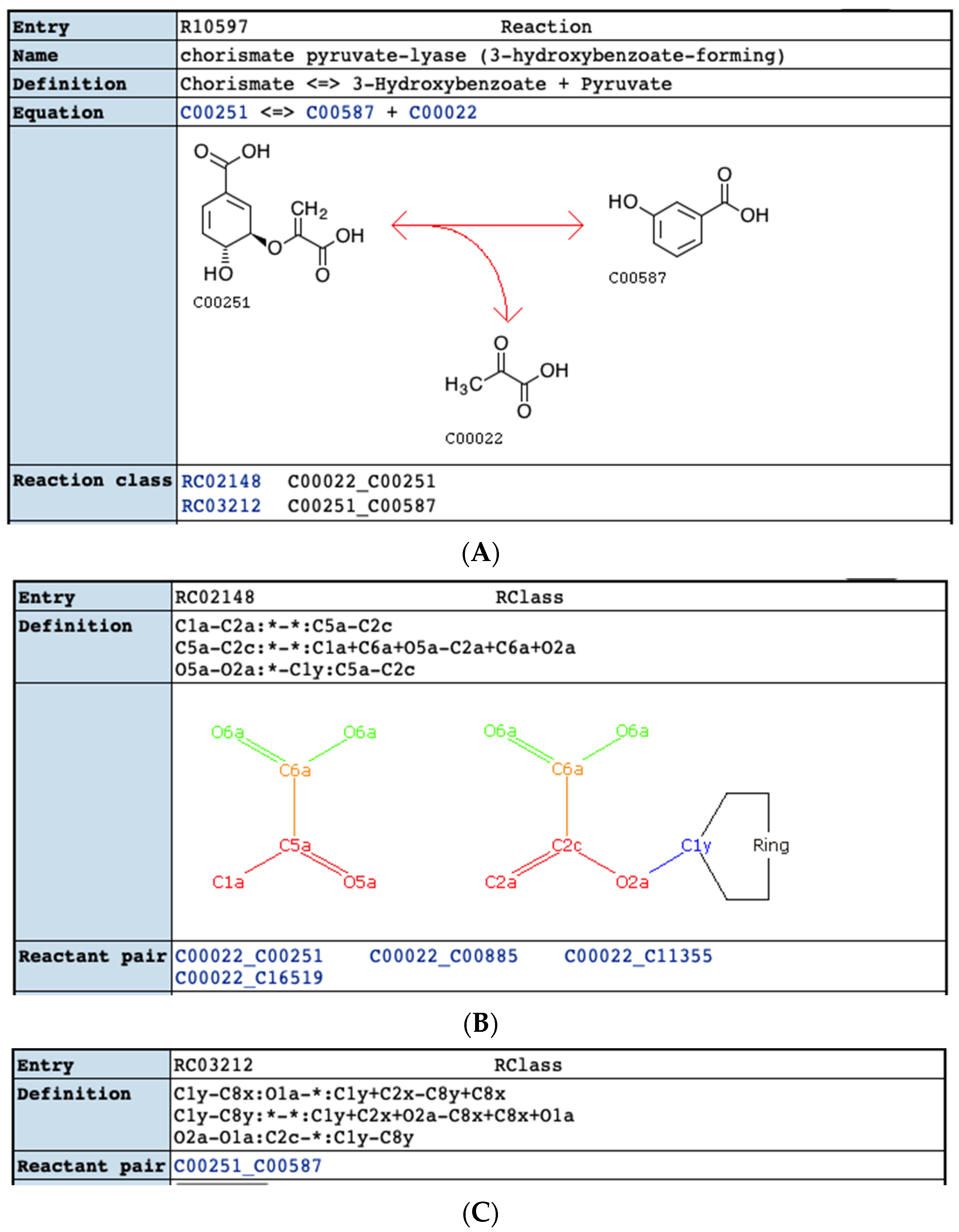
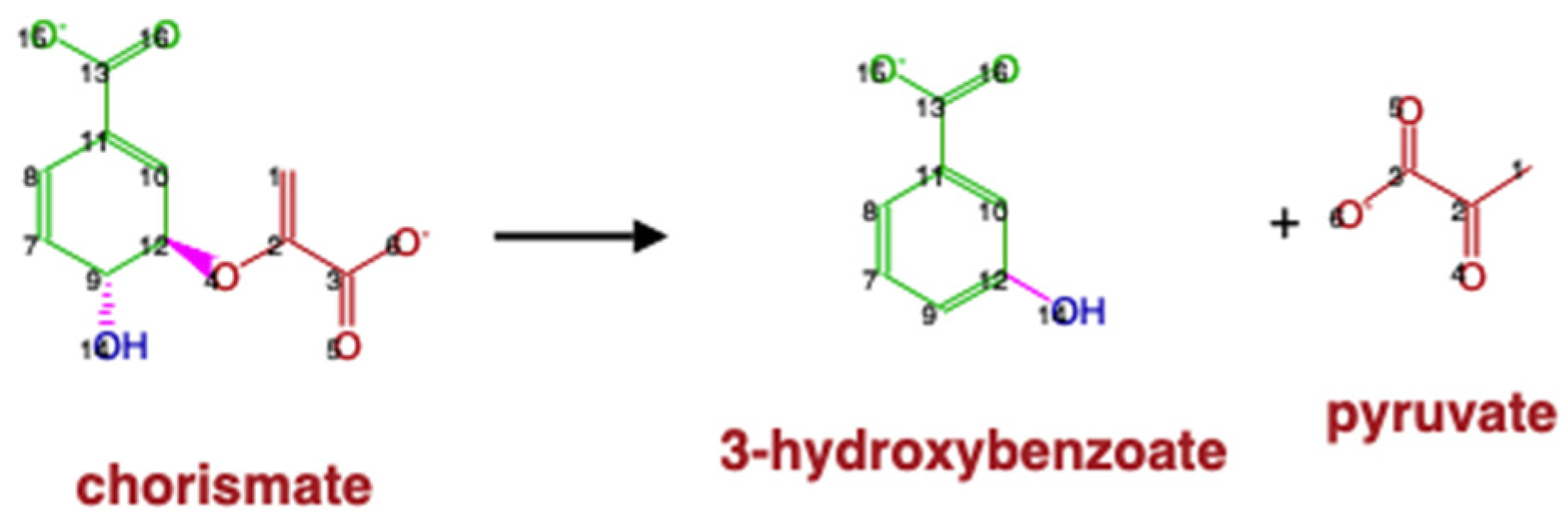
2.4. Comparison of KEGG RCLASS and RPAIR Data
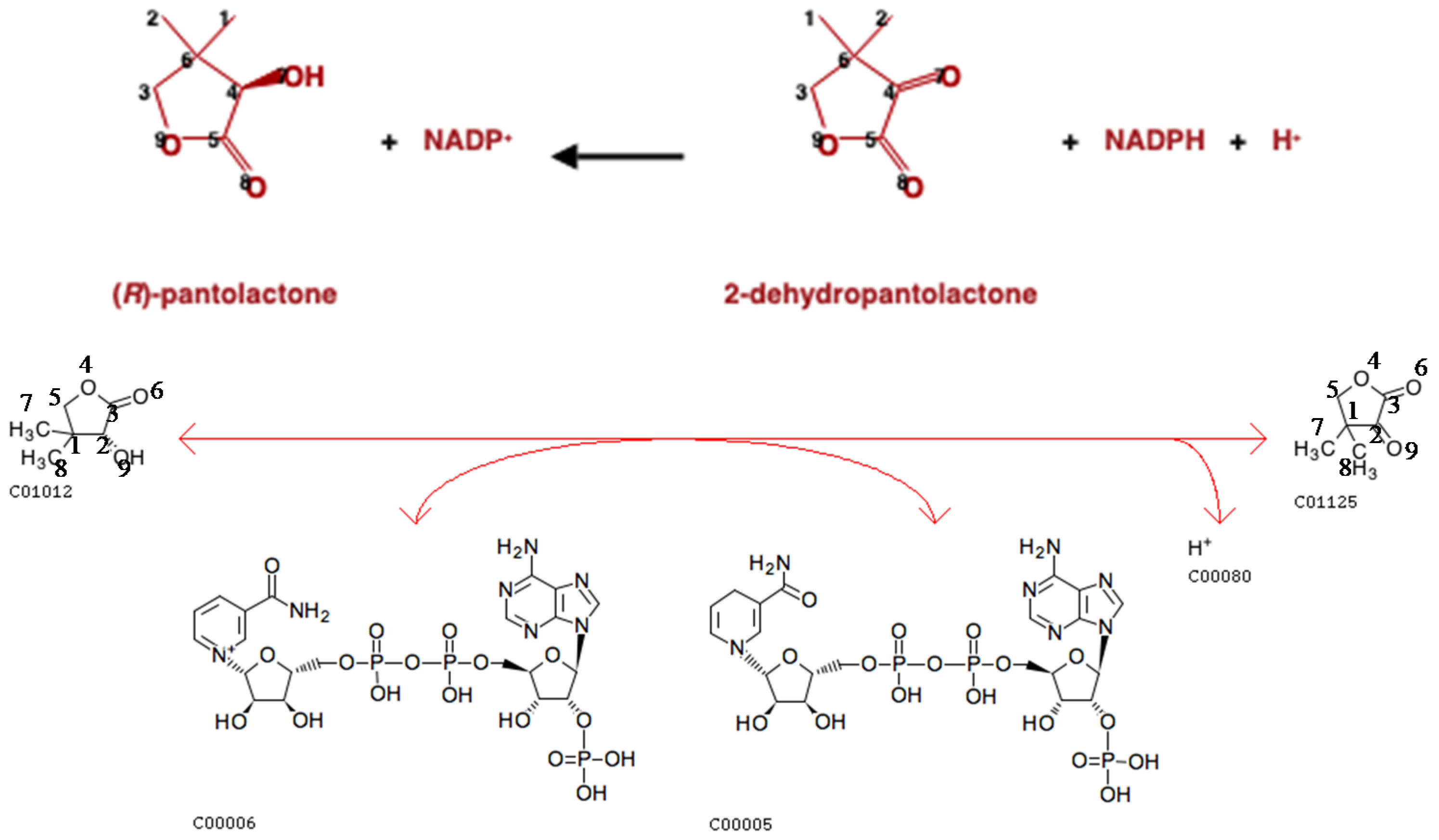
2.5. Evaluation of Atom Mappings between KEGG and MetaCyc Databases
3. Discussion
4. Materials and Methods
4.1. Compound and Metabolic Reaction Data
4.2. Curation of Molfile
4.3. Identification of Double Bond Stereochemistry
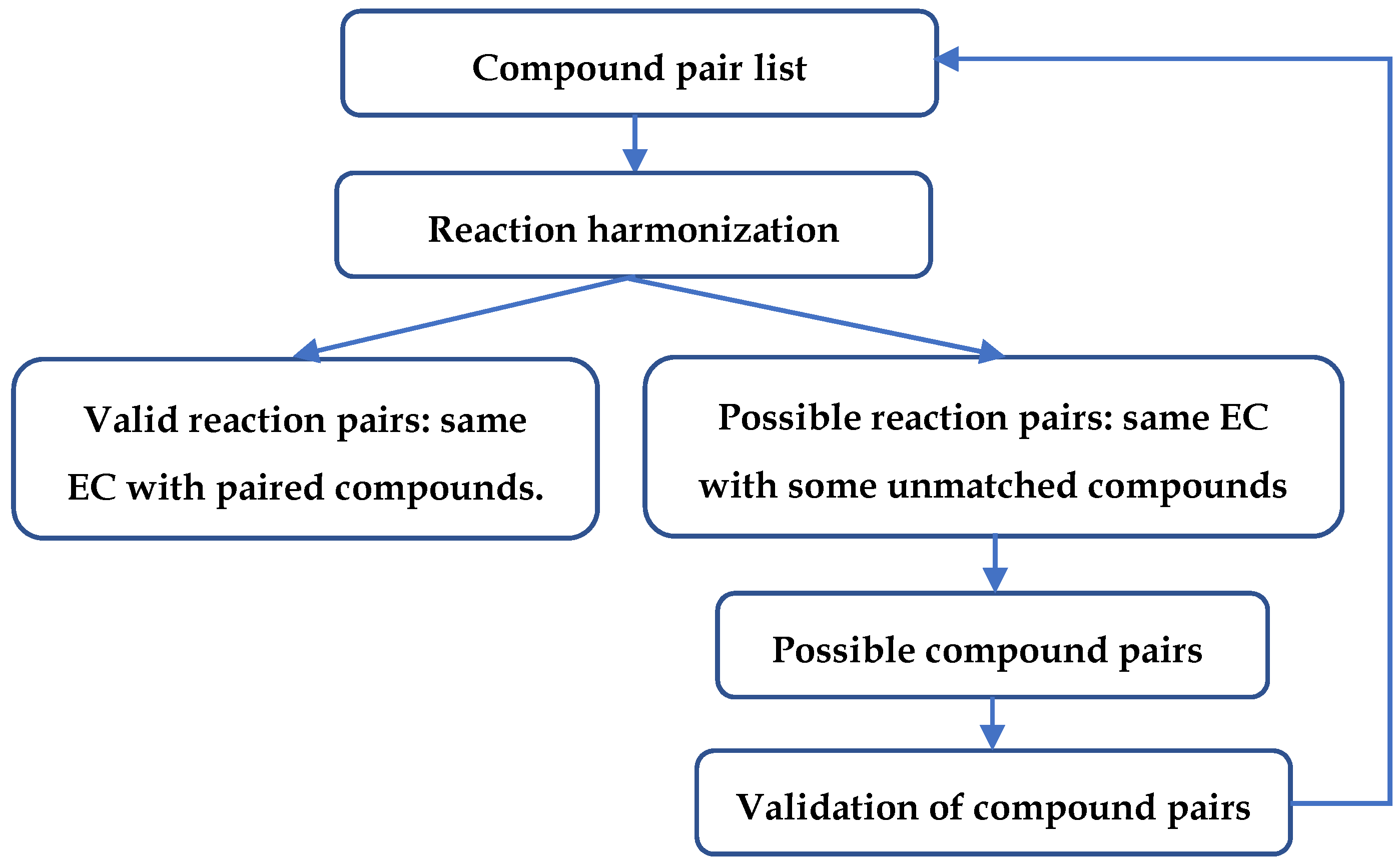
4.4. Flowchart of Steps in the Compound and Reaction Harmonization Process
4.5. Validation of Tautomers
4.6. Validation of Generic Compound Pairs of Compounds with Different Chemical Formula
4.7. Validation of Compound Pairs with Linear and Circular Representations
4.8. Parse of KEGG RCLASS RDM Patterns
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pham, N.; van Heck, R.G.A.; van Dam, J.C.J.; Schaap, P.J.; Saccenti, E.; Suarez-Diez, M. Consistency, Inconsistency and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Reconstruction of genome-scale human metabolic models using omics data. Integr. Biol. 2015, 7, 859–868. [Google Scholar] [CrossRef]
- Contreras, A.; Ribbeck, M.; Gutiérrez, G.D.; Cañon, P.M.; Mendoza, S.N.; Agosin, E. Mapping the Physiological Response of Oenococcus oeni to Ethanol Stress Using an Extended Genome-Scale Metabolic Model. Front. Microbiol. 2018, 9, 291. [Google Scholar] [CrossRef]
- Lee, D.S.; Burd, H.; Liu, J.; Almaas, E.; Wiest, O.; Barabasi, A.L.; Oltvai, Z.N.; Kapatral, V. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 2009, 191, 4015–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, K.R.; Akesson, M.; Nielsen, J. Use of genome-scale microbial models for metabolic engineering. Curr. Opin. Biotechnol. 2004, 15, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Radrich, K.; Tsuruoka, Y.; Dobson, P.; Gevorgyan, A.; Swainston, N.; Baart, G.; Schwartz, J.M. Integration of metabolic databases for the reconstruction of genome-scale metabolic networks. BMC Syst. Biol. 2010, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hua, Q. Applications of Genome-Scale Metabolic Models in Biotechnology and Systems Medicine. Front. Physiol. 2015, 6, 413. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Chokkathukalam, A.; Jankevics, A.; Burgess, K.E.; Breitling, R.; Barrett, M.P. Stable isotope-assisted metabolomics for network-wide metabolic pathway elucidation. Anal. Chem. 2012, 84, 8442–8447. [Google Scholar] [CrossRef]
- Fan, T.W.; Lorkiewicz, P.K.; Sellers, K.; Moseley, H.N.; Higashi, R.M.; Lane, A.N. Stable isotope-resolved metabolomics and applications for drug development. Pharm. Ther. 2012, 133, 366–391. [Google Scholar] [CrossRef] [Green Version]
- Antoniewicz, M.R. Methods and advances in metabolic flux analysis: A mini-review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef]
- Jin, H.; Moseley, H.N.B. Moiety modeling framework for deriving moiety abundances from mass spectrometry measured isotopologues. BMC Bioinform. 2019, 20, 524. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Moseley, H.N.B. Robust Moiety Model Selection Using Mass Spectrometry Measured Isotopologues. Metabolites 2020, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Young, J.D. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chokkathukalam, A.; Kim, D.H.; Barrett, M.P.; Breitling, R.; Creek, D.J. Stable isotope-labeling studies in metabolomics: New insights into structure and dynamics of metabolic networks. Bioanalysis 2014, 6, 511–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, T.; Travers, M.; Kothari, A.; Caspi, R.; Karp, P.D. A systematic comparison of the MetaCyc and KEGG pathway databases. BMC Bioinform. 2013, 14, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsburg, H. Caveat emptor: Limitations of the automated reconstruction of metabolic pathways in Plasmodium. Trends Parasitol. 2009, 25, 37–43. [Google Scholar] [CrossRef]
- Poolman, M.G.; Bonde, B.K.; Gevorgyan, A.; Patel, H.H.; Fell, D.A. Challenges to be faced in the reconstruction of metabolic networks from public databases. Syst. Biol. 2006, 153, 379–384. [Google Scholar] [CrossRef]
- Saha, R.; Chowdhury, A.; Maranas, C.D. Recent advances in the reconstruction of metabolic models and integration of omics data. Curr. Opin. Biotechnol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Ozsoyoglu, Z.M.; Ozsoyoglu, G. Matching metabolites and reactions in different metabolic networks. Methods 2014, 69, 282–297. [Google Scholar] [CrossRef]
- van Heck, R.G.; Ganter, M.; Martins Dos Santos, V.A.; Stelling, J. Efficient Reconstruction of Predictive Consensus Metabolic Network Models. PLoS Comput. Biol. 2016, 12, e1005085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI—The worldwide chemical structure identifier standard. J. Cheminform. 2013, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. Memote: A community driven effort towards a standardized genome-scale metabolic model test suite. Nat Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Mitchell, J.M.; Moseley, H.N.B. Atom Identifiers Generated by a Neighborhood-Specific Graph Coloring Method Enable Compound Harmonization across Metabolic Databases. Metabolites 2020, 10, 368. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Fan, T.W.-M.; Lane, A.N.; Moseley, H.N.B. Development and in silico evaluation of large-scale metabolite identification methods using functional group detection for metabolomics. Front. Genet. 2014, 5, 237. [Google Scholar] [CrossRef] [Green Version]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.J.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Hadadi, N.; Hafner, J.; Shajkofci, A.; Zisaki, A.; Hatzimanikatis, V. ATLAS of Biochemistry: A Repository of All Possible Biochemical Reactions for Synthetic Biology and Metabolic Engineering Studies. ACS Synth. Biol. 2016, 5, 1155–1166. [Google Scholar] [CrossRef]
- Frainay, C.; Schymanski, E.L.; Neumann, S.; Merlet, B.; Salek, R.M.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Barrett, A.J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: Corrections and additions (1997). Eur. J. Biochem. 1997, 250, 1–6. [Google Scholar] [PubMed]
- McDonald, A.G.; Boyce, S.; Tipton, K.F. ExplorEnz: The primary source of the IUBMB enzyme list. Nucleic Acids Res. 2008, 37 (Suppl. S1), D593–D597. [Google Scholar] [CrossRef]
- Kotera, M.; Okuno, Y.; Hattori, M.; Goto, S.; Kanehisa, M. Computational Assignment of the EC Numbers for Genomic-Scale Analysis of Enzymatic Reactions. J. Am. Chem. Soc. 2004, 126, 16487–16498. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef] [PubMed]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2018, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, A.L.; Leal, J.P.; Falcao, A. Automated Identification and Classification of Stereochemistry: Chirality and Double Bond Stereoisomerism. arXiv 2013, arXiv:abs/1303.1724. [Google Scholar]

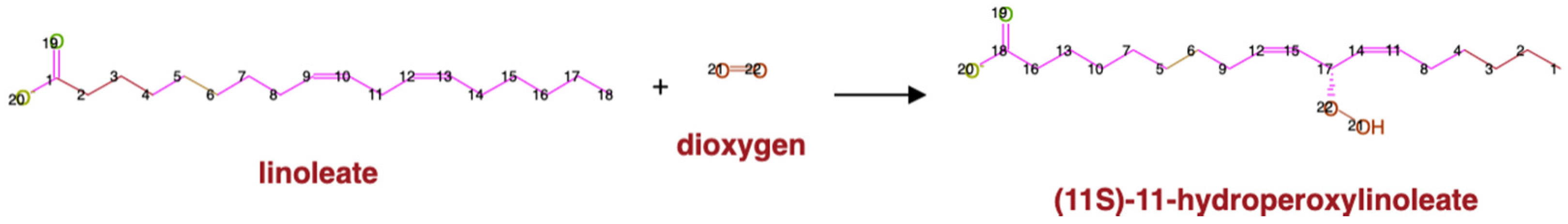

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Type | KEGG | MetaCyc |
|---|---|---|
| specific compounds | 16,529 (91.98%) | 15,859 (78.28%) |
| generic compounds | 1441 (8.02%) | 4400 (21.72%) |
| Total | 17,970 (100%) | 20,259 (100%) |
| Reaction Type | KEGG | MetaCyc |
|---|---|---|
| specific reactions (4-leveled EC) | 6780 (75.26%) | 6397 (49.93%) |
| specific reactions (3-leveled EC) | 886 (9.83%) | 2022 (15.78%) |
| generic reactions (4-leveled EC) | 1244 (13.81%) | 3572 (27.88%) |
| generic reactions (3-leveled EC) | 99 (1.10%) | 822 (6.42%) |
| Total | 9009 (100%) | 12,813 (100%) |
| Database | Incomplete Reaction | Incorrect Atom Mappings | Incomplete Atom Mappings |
|---|---|---|---|
| KEGG | 772 (7.53%) | 0 | 7213 (70.36%) |
| MetaCyc | 0 | 54 (0.37%) | 6130 (41.87%) |
| Relationship Type | Count |
|---|---|
| equivalence | 3636 (27.99%) |
| generic-specific | 1712 (13.18%) |
| loose | 7642 (58.83%) |
| Total | 12,990 (100%) |
| Relationship Type | Count |
|---|---|
| equivalence | 126 (4.72%) |
| generic-specific | 2543 (95.28%) |
| loose | 0 |
| Total | 2669 (100%) |
| Relationship Type | Count |
|---|---|
| equivalence | 20 (44.44%) |
| generic-specific | 0 |
| loose | 25 (55.56%) |
| Total | 45 (100%) |
| Compound Pair Type | Count |
|---|---|
| specific compound pairs | 12,990 (82.72%) |
| generic compound pairs | 2669 (16.99%) |
| changeable compound pairs | 45 (0.29%) |
| Total | 15,704 (100%) |
| Relationship Type | Count |
|---|---|
| equivalence | 718 (24.00%) |
| generic-specific | 68 (2.27%) |
| loose | 2205 (73.72%) |
| Total | 2991 (100%) |
| Relationship Type | Count |
|---|---|
| equivalence | 29 (6.03%) |
| generic-specific | 344 (71.51%) |
| loose | 108 (22.45%) |
| Total | 481 (100%) |
| Relationship Type | Count |
|---|---|
| equivalence | 49 (12.76%) |
| generic-specific | 96 (25.00%) |
| loose | 239 (62.24%) |
| Total | 384 (100%) |
| Reaction Pair Type | Count |
|---|---|
| specific | 2991 (77.57%) |
| generic | 481 (12.47%) |
| loose EC | 384 (9.96%) |
| Total | 3856 (100%) |
| Condition | Count |
|---|---|
| same atom mappings | 8017 (86.1%) |
| inconsistent atom mappings | 1294 (13.9%) |
| Total | 9311 (100%) |
| Condition | Count |
|---|---|
| same atom mappings | 8754 (94.02%) |
| inconsistent atom mappings | 557 (5.98%) |
| Total | 9311 (100%) |
| Condition | Count |
|---|---|
| same atom mappings | 2685 (88.0%) |
| inconsistent atom mappings | 366 (12.0%) |
| Total | 3051 (100%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Moseley, H.N.B. Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases. Metabolites 2021, 11, 431. https://doi.org/10.3390/metabo11070431
Jin H, Moseley HNB. Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases. Metabolites. 2021; 11(7):431. https://doi.org/10.3390/metabo11070431
Chicago/Turabian StyleJin, Huan, and Hunter N. B. Moseley. 2021. "Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases" Metabolites 11, no. 7: 431. https://doi.org/10.3390/metabo11070431
APA StyleJin, H., & Moseley, H. N. B. (2021). Hierarchical Harmonization of Atom-Resolved Metabolic Reactions across Metabolic Databases. Metabolites, 11(7), 431. https://doi.org/10.3390/metabo11070431