
A Single LC-MS/MS Analysis to Quantify CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs

 ,
, 
Abstract
1. Introduction
2. Results
2.1. The MRM MS/MS Method Can Detect Both CoA Biosynthetic Intermediates and Short-Chain Acyl-CoAs
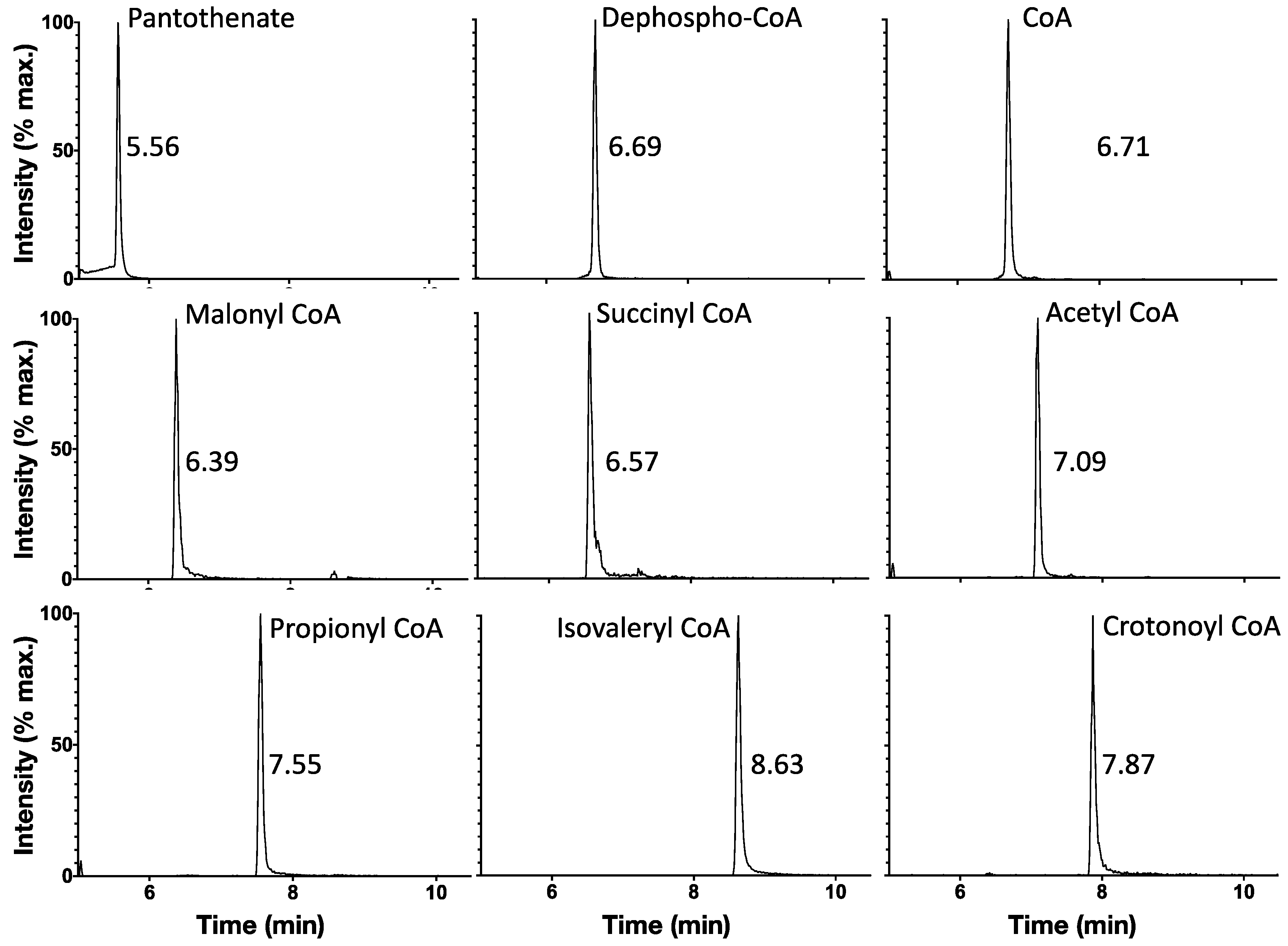
2.2. Ion-Pairing UHPLC Chromatography Produces Well-Separated Peaks for CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs
2.3. The Method Displays a Linear Detection of Analytes across a Wide Concentration Range with Sensitive Lower Limits of Detection and Quantitation
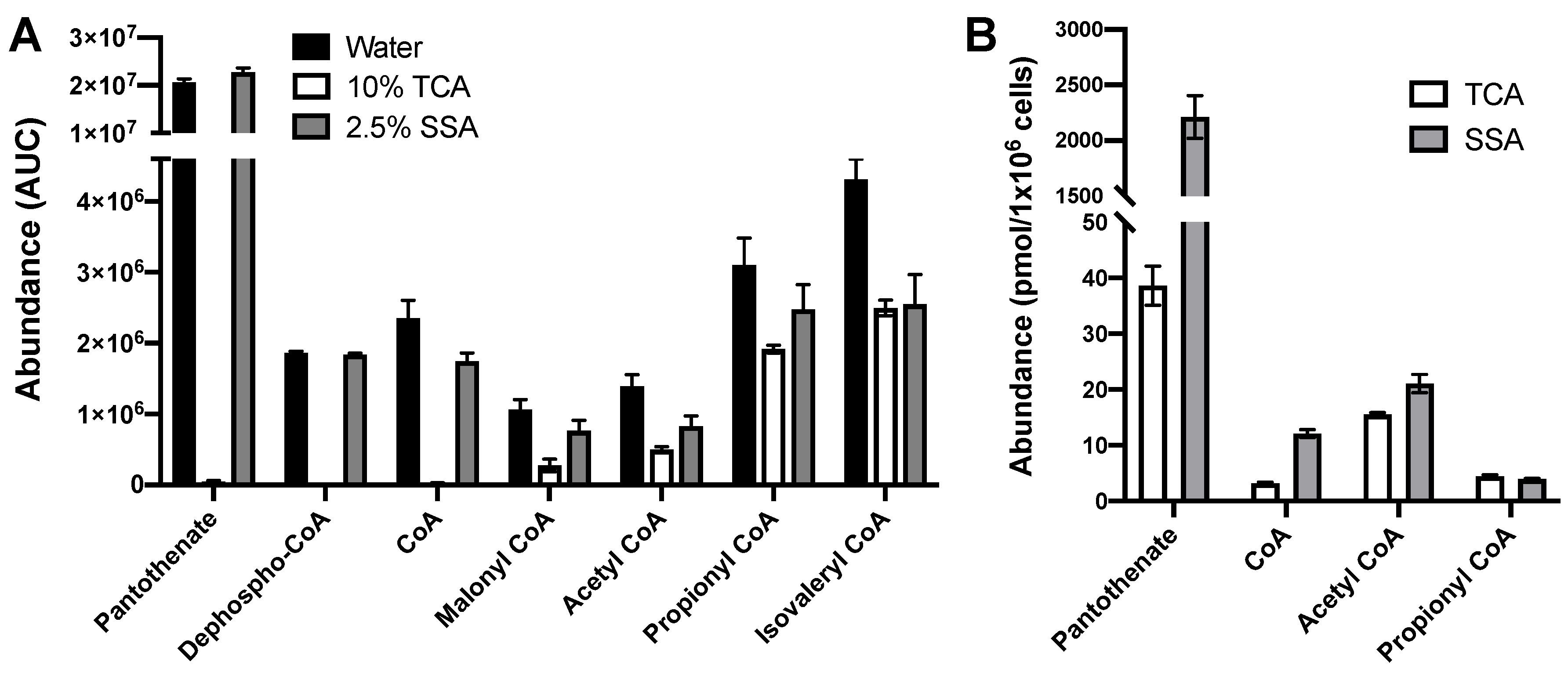
2.4. Extraction with 2.5% SSA Is Suitable for Analysis of Acyl CoAs and CoA Biosynthetic Intermediates
2.5. The Sample Preparation and LC-MS/MS Method Demonstrates the Minimal Matrix Effect and Preserves Accuracy
2.6. The Method Exhibits Acceptable Precision and Accuracy Parameters for Measured Analytes Across Their Entire Linear Ranges
2.7. The Method Detects Expected Perturbations in Levels of CoA and CoA Esters
2.8. The Method Detects Changes in the CoA Biosynthetic Pathway and Short-Chain Acyl CoA Species in IL-4-Polarized Macrophages Treated with Excess Etomoxir
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Lines and Culture
4.2.1. Snap-Freezing of Cell Pellets
4.2.2. Extraction of Pathway Intermediates and CoA Species from Cell Pellets with 5-Sulfosalicylic Acid (SSA) or Trichloroacetic Acid (TCA)
4.3. ESI LC-MS/MS Analytical Method
4.3.1. Liquid Chromatography
4.3.2. Mass Spectrometry
4.3.3. Data Analysis
4.4. Validation of Sample Preparation and LC-MS/MS Methods
4.4.1. Linearity, Lower Limit of Detection (LLOD), and Lower Limit of Quantitation (LLOQ)
4.4.2. Matrix Effect and Comparative Accuracy
4.4.3. Precision and Accuracy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonardi, R.; Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.D.; Wang, H.; Kekuda, R.; Fujita, T.; Fei, Y.-J.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V. Cloning and Functional Expression of a cDNA Encoding a Mammalian Sodium-dependent Vitamin Transporter Mediating the Uptake of Pantothenate, Biotin, and Lipoate. J. Biol. Chem. 1998, 273, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Quick, M.; Shi, L. The Sodium/Multivitamin Transporter: A multipotent system with therapeutic implications. Vitam. Horm. 2015, 98, 63–100. [Google Scholar] [CrossRef] [PubMed]
- Robishaw, J.D.; Neely, J.R. Pantothenate kinase and control of CoA synthesis in heart. Am. J. Physiol. 1984, 246, H532–H541. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.G.; Bartlett, M.G. Chromatographic methods for the determination of acyl-CoAs. Anal. Methods 2018, 10, 5252–5264. [Google Scholar] [CrossRef]
- Vessey, D.A.; Kelley, M.; Warren, R.S. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim. Biophys. Acta 1999, 1428, 455–462. [Google Scholar] [CrossRef]
- Gauthier, N.; Wu, J.W.; Wang, S.P.; Allard, P.; Mamer, O.A.; Sweetman, L.; Moser, A.B.; Kratz, L.; Álvarez, F.; Robitaille, Y.; et al. A Liver-Specific Defect of Acyl-CoA Degradation Produces Hyperammonemia, Hypoglycemia and a Distinct Hepatic Acyl-CoA Pattern. PLoS ONE 2013, 8, e60581. [Google Scholar] [CrossRef]
- Venco, P.; Dusi, S.; Valletta, L.; Tiranti, V. Alteration of the coenzyme A biosynthetic pathway in neurodegeneration with brain iron accumulation syndromes. Biochem. Soc. Trans. 2014, 42, 1069–1074. [Google Scholar] [CrossRef]
- Gregory, A.; Hayflick, S.J. Pantothenate Kinase-Associated Neurodegeneration. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Covarrubias, A.; Aksoylar, H.I.; Yu, J.; Snyder, N.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503.e7. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Chu, D.B.; Marx, H.; Sauer, M.; Hann, S.; Koellensperger, G. LC-MS/MS-based analysis of coenzyme A and short-chain acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 2015, 407, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Pham, U.; Gout, I. Methods for measuring CoA and CoA derivatives in biological samples. Biochem. Soc. Trans. 2014, 42, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.A.D.; Choi, S.-Y.; Wurtele, E.; Nikolau, B.J. Quantitative analysis of short-chain acyl-coenzymeAs in plant tissues by LC–MS–MS electrospray ionization method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Snyder, N.; Basu, S.S.; Worth, A.J.; Mesaros, C.; Blair, I.A. Metabolism of propionic acid to a novel acyl-coenzyme A thioester by mammalian cell lines and platelets. J. Lipid Res. 2015, 56, 142–150. [Google Scholar] [CrossRef]
- Gao, L.; Chiou, W.; Tang, H.; Cheng, X.; Camp, H.S.; Burns, D.J. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 853, 303–313. [Google Scholar] [CrossRef]
- Park, J.W.; Jung, W.S.; Park, S.R.; Park, B.C.; Yoon, Y.J. Analysis of intracellular short organic acid-coenzyme A esters from actinomycetes using liquid chromatography-electrospray ionization-mass spectrometry. J. Mass Spectrom. 2007, 42, 1136–1147. [Google Scholar] [CrossRef]
- Hayashi, O.; Satoh, K. Determination of Acetyl-CoA and Malonyl-CoA in Germinating Rice Seeds Using the LC-MS/MS Technique. Biosci. Biotechnol. Biochem. 2006, 70, 2676–2681. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Minkler, P.E.; Kerner, J.; Ingalls, S.T.; Hoppel, C.L. Novel isolation procedure for short-, medium-, and long-chain acyl-coenzyme A esters from tissue. Anal. Biochem. 2008, 376, 275–276. [Google Scholar] [CrossRef]
- Basu, S.S.; Blair, I.A. SILEC: A protocol for generating and using isotopically labeled coenzyme A mass spectrometry standards. Nat. Protoc. 2011, 7, 1–11. [Google Scholar] [CrossRef]
- Basu, S.S.; Mesaros, C.; Gelhaus, S.L.; Blair, I.A. Stable Isotope Labeling by Essential Nutrients in Cell Culture for Preparation of Labeled Coenzyme A and Its Thioesters. Anal. Chem. 2011, 83, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, G.W.; Fenton, T.W.; Thompson, J.R. Effect of protein precipitating agents on the recovery of plasma free amino acids. Can. J. Anim. Sci. 1991, 71, 953–957. [Google Scholar] [CrossRef]
- Berrueta, L.A.; Gallo, B.; Vicente, F. A review of solid phase extraction: Basic principles and new developments. Chromatographia 1995, 40, 474–483. [Google Scholar] [CrossRef]
- Abrankó, L.; Williamson, G.; Gardner, S.; Kerimi, A. Comprehensive quantitative analysis of fatty-acyl-Coenzyme A species in biological samples by ultra-high performance liquid chromatography–tandem mass spectrometry harmonizing hydrophilic interaction and reversed phase chromatography. J. Chromatogr. A 2018, 1534, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Palladino, A.A.; Chen, J.; Kallish, S.; Stanley, C.; Bennett, M.J. Measurement of tissue acyl-CoAs using flow-injection tandem mass spectrometry: Acyl-CoA profiles in short-chain fatty acid oxidation defects. Mol. Genet. Metab. 2012, 107, 679–683. [Google Scholar] [CrossRef]
- Chemdata: Coenzyme A. Available online: https://www.drugfuture.com/chemdata/coenzyme-a.html (accessed on 25 April 2021).
- Pubchem Compound Summary: N,N-dimethybutylamine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/N_N-Dimethylbutylamine (accessed on 25 April 2021).
- Basiri, B.; Murph, M.M.; Bartlett, M.G. Assessing the Interplay between the Physicochemical Parameters of Ion-Pairing Reagents and the Analyte Sequence on the Electrospray Desorption Process for Oligonucleotides. J. Am. Soc. Mass Spectrom. 2017, 28, 1647–1656. [Google Scholar] [CrossRef]
- Demoz, A.; Garras, A.; Asiedu, D.K.; Netteland, B.; Berge, R.K. Rapid method for the separation and detection of tissue short-chain coenzyme A esters by reversed-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1995, 667, 148–152. [Google Scholar] [CrossRef]
- Khan, K.; Blaak, E.; Elia, M. Quantifying intermediary metabolites in whole blood after a simple deproteinization step with sulfosalicylic acid. Clin. Chem. 1991, 37, 728–733. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services: Food and Drug Administration Center for Drug Evaluation and Research and Center for Veterinary Medicine. Guidance for Industry, Bioanalytical Method Validation. 2001. Available online: http://www.fda.gov/cder/guidance/index.htm (accessed on 25 April 2021).
- Smith, G. European Medicines Agency guideline on bioanalytical method validation: What more is there to say? Bioanalysis 2012, 4, 865–868. [Google Scholar] [CrossRef]
- Sharma, L.K.; Subramanian, C.; Yun, M.-K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef]
- Ulrich, R.G.; Bacon, J.A.; Brass, E.P.; Cramer, C.T.; Petrella, D.K.; Sun, E.L. Metabolic, idiosyncratic toxicity of drugs: Overview of the hepatic toxicity induced by the anxiolytic, panadiplon. Chem. Biol. Interact. 2001, 134, 251–270. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef]
- Rock, C.O.; Karim, M.A.; Zhang, Y.-M.; Jackowski, S. The murine pantothenate kinase (Pank1) gene encodes two differentially regulated pantothenate kinase isozymes. Gene 2002, 291, 35–43. [Google Scholar] [CrossRef]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme a metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome Sequence Reveals Mutations in CoA Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Leonardi, R.; Zhang, Y.-M.; Rehg, J.E.; Jackowski, S. Germline Deletion of Pantothenate Kinases 1 and 2 Reveals the Key Roles for CoA in Postnatal Metabolism. PLoS ONE 2012, 7, e40871. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.O.; Calder, R.B.; Karim, M.A.; Jackowski, S. Pantothenate Kinase Regulation of the Intracellular Concentration of Coenzyme A. J. Biol. Chem. 2000, 275, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, W.-Y.; Williams, K.J.; Su, B.; Bensinger, S.J. Profiling of mouse macrophage lipidome using direct infusion shotgun mass spectrometry. STAR Protoc. 2021, 2, 100235. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.L. CLSI C62-A: A New Standard for Clinical Mass Spectrometry. Clin. Chem. 2016, 62, 24–29. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Acyl Group Formula | Acyl Group Structure | Parent (m/z) | Daughter 1 (m/z) | Daughter 2 (m/z) |
| CoA | H | H | 768.1 | 261.1 | 428.1 |
| Acetyl CoA | COCH3 |  | 810.1 | 303.1 | 428.1 |
| Propionyl CoA | COCH2CH3 |  | 824.1 | 317.1 | 428.1 |
| Isovaleryl CoA | COCH2(CH3)2 |  | 852.1 | 345.1 | 428.1 |
| Malonyl CoA | COCH2CO2H |  | 854.1 | 347.1 | 428.1 |
| Succinyl CoA | CO(CH2)2CO2H |  | 868.1 | 361.1 | 428.1 |
| Compoud Name | Chemical Formula | Compound Structure | Parent (m/z) | Daughter 1 (m/z) | Daughter 2 (m/z) |
| Pantothenate | C9H17NO5 |  | 218.0 | 88.0 | 145.8 |
| Dephospho-CoA | C21H35N7O13P2S |  | 688.1 | 261.1 | 348.1 |
| Time (min) | %A | %B | Notes |
|---|---|---|---|
| 0 | 98 | 2 | Divert to waste (0–5.5 min) |
| 1.5 | 98 | 2 | |
| 9 | 50 | 50 | Divert to MS (5.5–10.5 min) |
| 9.5 | 2 | 98 | |
| 14.5 | 2 | 98 | Divert to waste (10.5–22 min) |
| 15 | 98 | 2 | |
| 22 | 98 | 2 |
| Compound | Linear Regression | r | LLOD (pmol) | LLOQ (pmol) |
|---|---|---|---|---|
| Pantothenate | Y = 0.00264x + 0.0307 | 0.98 | 1 | 7.4 |
| Dephospho-CoA | Y = 0.00481x + 0.0892 | 0.97 | 0.4 | 3.7 |
| CoA | Y = 0.00546x − 0.0196 | 0.97 | 1 | 3.7 |
| Malonyl CoA | Y = 0.00221x − 0.00851 | 0.99 | 3 | 3.7 |
| Succinyl CoA | Y = 0.00095x − 0.0011 | 0.95 | 1 | 7.4 |
| Acetyl CoA | Y = 0.00461x − 0.0163 | 0.97 | 1 | 3.7 |
| Propionyl CoA | Y = 0.00660x + 0.0367 | 0.99 | 2 | 3.7 |
| Isovaleryl CoA | Y = 0.00864x − 0.1853 | 0.99 | 1 | 7.4 |
| Standards (pmol) | Calculated Concentration (pmol/1 × 106 cells) | SEM | Precision CV (%) | Accuracy (% of Nominal Value) | |
|---|---|---|---|---|---|
| Pantothenate | 0 | 969.15 | 7.74 | 1.60 | NA |
| 62.5 | 1231.50 | 48.44 | 7.87 | 119.37 | |
| 250 | 1421.00 | 78.83 | 11.10 | 116.56 | |
| 1000 | 2244.50 | 31.47 | 2.80 | 113.98 | |
| Dephospho-CoA | 0 | 1.68 | 0.08 | 9.78 | NA |
| 62.5 | 57.39 | 4.78 | 16.64 | 89.41 | |
| 250 | 228.81 | 23.02 | 20.12 | 90.91 | |
| 1000 | 961.96 | 24.63 | 5.12 | 96.03 | |
| CoA | 0 | 49.98 | 4.67 | 18.68 | NA |
| 62.5 | 143.03 | 4.81 | 6.73 | 127.16 | |
| 250 | 277.50 | 16.61 | 11.97 | 92.51 | |
| 1000 | 865.48 | 7.02 | 1.62 | 82.43 | |
| Acetyl CoA | 0 | 65.67 | 1.81 | 5.52 | NA |
| 62.5 | 127.05 | 8.85 | 13.94 | 99.13 | |
| 250 | 356.48 | 14.96 | 8.40 | 112.93 | |
| 1000 | 1402.79 | 32.15 | 4.58 | 131.63 | |
| Propionyl CoA | 0 | 7.95 | 0.20 | 5.03 | NA |
| 62.5 | 75.03 | 5.98 | 15.93 | 106.49 | |
| 250 | 277.52 | 4.06 | 2.92 | 107.59 | |
| 1000 | 1132.35 | 59.58 | 10.52 | 112.34 | |
| Isovaleryl CoA | 0 | 3.17 | 0.21 | 13.19 | NA |
| 62.5 | 67.53 | 6.33 | 18.76 | 102.82 | |
| 250 | 314.52 | 4.34 | 2.76 | 124.23 | |
| 1000 | 1383.19 | 50.47 | 7.30 | 137.88 | |
| Malonyl CoA | 0 | 2.45 | 0.18 | 14.84 | NA |
| 62.5 | 62.03 | 2.94 | 9.48 | 95.50 | |
| 250 | 265.98 | 20.15 | 15.15 | 105.36 | |
| 1000 | 1077.85 | 69.21 | 12.84 | 107.52 | |
| Succinyl CoA | 0 | 208.61 | 11.79 | 11.31 | NA |
| 62.5 | 295.38 | 22.62 | 15.32 | 108.96 | |
| 250 | 507.42 | 25.24 | 9.95 | 110.64 | |
| 1000 | 1279.86 | 71.49 | 11.17 | 105.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, A.E.; Arias, N.J.; Acevedo, A.; Reddy, S.T.; Divakaruni, A.S.; Meriwether, D. A Single LC-MS/MS Analysis to Quantify CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs. Metabolites 2021, 11, 468. https://doi.org/10.3390/metabo11080468
Jones AE, Arias NJ, Acevedo A, Reddy ST, Divakaruni AS, Meriwether D. A Single LC-MS/MS Analysis to Quantify CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs. Metabolites. 2021; 11(8):468. https://doi.org/10.3390/metabo11080468
Chicago/Turabian StyleJones, Anthony E., Nataly J. Arias, Aracely Acevedo, Srinivasa T. Reddy, Ajit S. Divakaruni, and David Meriwether. 2021. "A Single LC-MS/MS Analysis to Quantify CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs" Metabolites 11, no. 8: 468. https://doi.org/10.3390/metabo11080468
APA StyleJones, A. E., Arias, N. J., Acevedo, A., Reddy, S. T., Divakaruni, A. S., & Meriwether, D. (2021). A Single LC-MS/MS Analysis to Quantify CoA Biosynthetic Intermediates and Short-Chain Acyl CoAs. Metabolites, 11(8), 468. https://doi.org/10.3390/metabo11080468