
Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma

{kind=link}
Abstract
:1. Introduction
2. Lipid Uptake and Metabolism in Normal Proximal Tubule Cells
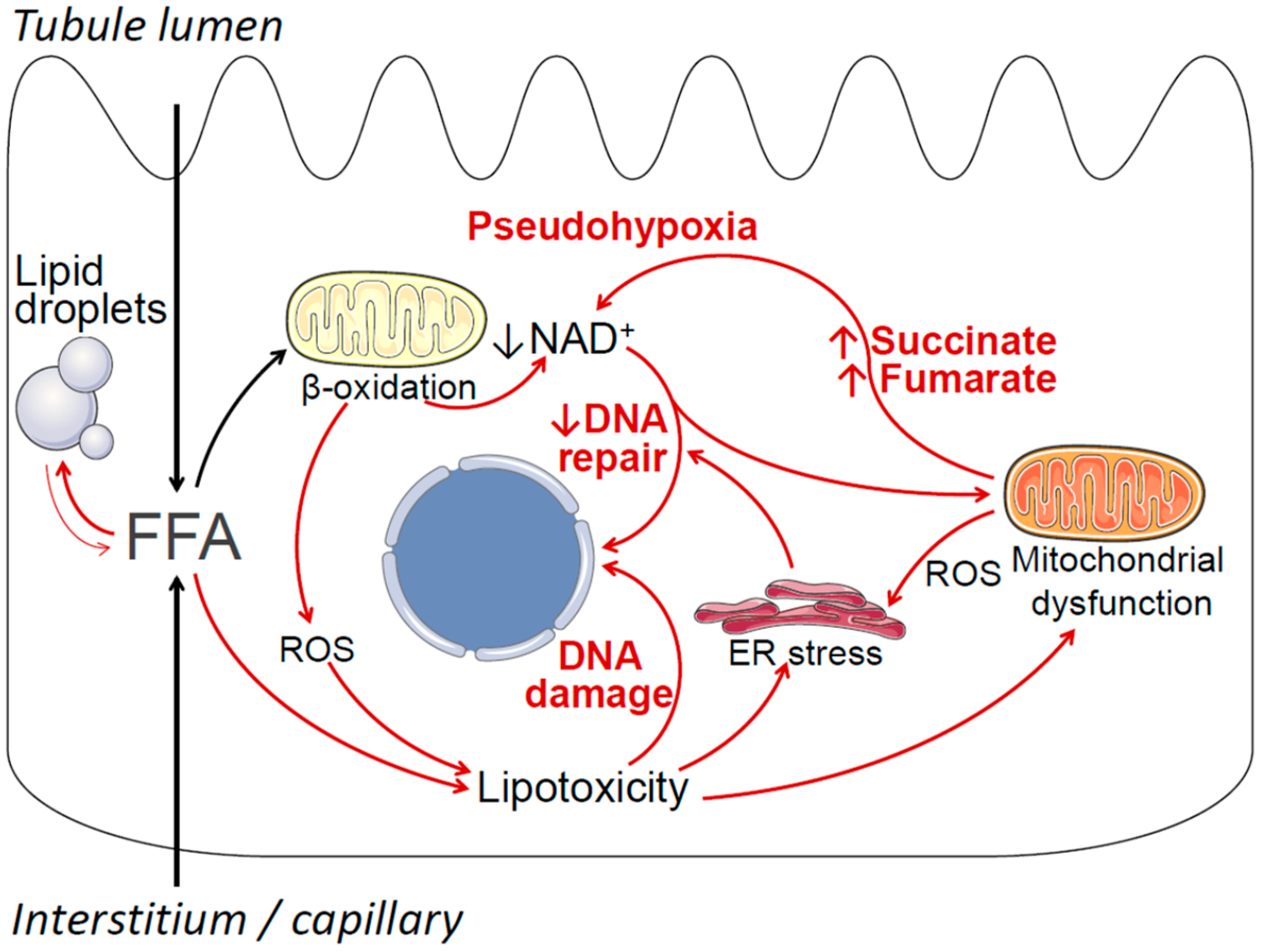
3. Lipid Metabolic Disturbances in Non-Cancerous Proximal Tubule Cells
4. Lipid Metabolic Reprogramming in ccRCC
5. Exploring the Link between Obesity and Renal Cell Carcinoma
6. Alterations in Renal Lipid Metabolism: A Potential Link between Obesity and ccRCC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017, National Cancer Institute: Bethesda, MD, USA. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 12 March 2021).
- Buttner, F.; Winter, S.; Rausch, S.; Reustle, A.; Kruck, S.; Junker, K.; Stenzl, A.; Agaimy, A.; Hartmann, A.; Bedke, J.; et al. Survival prediction of clear cell renal cell carcinoma based on gene expression similarity to the proximal tubule of the nephron. Eur. Urol. 2015, 68, 1016–1020. [Google Scholar] [CrossRef]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of renal cell carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Ricketts, C.J.; de Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Chow, W.H.; Dong, L.M.; Devesa, S.S. Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Reuter, V.E.; Tickoo, S.K. Differential diagnosis of renal tumours with clear cell histology. Pathology 2010, 42, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobulescu, I.A.; Lotan, Y.; Zhang, J.; Rosenthal, T.R.; Rogers, J.T.; Adams-Huet, B.; Sakhaee, K.; Moe, O.W. Triglycerides in the human kidney cortex: Relationship with body size. PLoS ONE 2014, 9, e101285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoo, T.; Clark, H.R.; Pedrosa, I.; Yuan, Q.; Dimitrov, I.; Zhang, Y.; Lingvay, I.; Beg, M.S.; Bobulescu, I.A. Quantification of renal steatosis in type II diabetes mellitus using dixon-based MRI. J. Magn. Reson. Imaging 2016, 44, 1312–1319. [Google Scholar] [CrossRef] [Green Version]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Takahashi, M.; Hori, M.; Ishigamori, R.; Mutoh, M.; Imai, T.; Nakagama, H. Fatty pancreas: A possible risk factor for pancreatic cancer in animals and humans. Cancer Sci. 2018, 109, 3013–3023. [Google Scholar] [CrossRef]
- Meyer, C.; Nadkarni, V.; Stumvoll, M.; Gerich, J. Human kidney free fatty acid and glucose uptake: Evidence for a renal glucose-fatty acid cycle. Am. J. Physiol. 1997, 273 Pt 1, E650–E654. [Google Scholar] [CrossRef]
- Nieth, H.; Schollmeyer, P. Substrate-utilization of the human kidney. Nature 1966, 209, 1244–1245. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Okamura, D.M.; Lopez-Guisa, J.M.; Koelsch, K.; Collins, S.; Eddy, A.A. Atherogenic scavenger receptor modulation in the tubulointerstitium in response to chronic renal injury. Am. J. Physiol. Renal Physiol. 2007, 293, F575–F585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 mediated fatty acid-induced podocyte apoptosis via oxidative stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshbach, M.L.; Weisz, O.A. Receptor-mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birn, H.; Christensen, E.I. Renal albumin absorption in physiology and pathology. Kidney Int. 2006, 69, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Cabral, P.D.; Schilling, W.P.; Schmidt, Z.W.; Uddin, A.N.; Gingras, A.; Madhavan, S.M.; Garvin, J.L.; Schelling, J.R. Kidney proximal tubule lipoapoptosis is regulated by fatty acid transporter-2 (FATP2). J. Am. Soc. Nephrol. 2018, 29, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Fatty Acid Synthase. Available online: https://www.proteinatlas.org/ENSG00000169710-FASN (accessed on 22 March 2021).
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar] [CrossRef]
- Weinberg, J.M. Lipotoxicity. Kidney Int. 2006, 70, 1560–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahba, I.M.; Mak, R.H. Obesity and obesity-initiated metabolic syndrome: Mechanistic links to chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 550–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, X.Z.; Varghese, Z.; Moorhead, J.F. An update on the lipid nephrotoxicity hypothesis. Nat. Rev. Nephrol. 2009, 5, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, Z.; Proctor, G.; Moskowitz, S.; Liebman, S.E.; Rogers, T.; Lucia, M.S.; Li, J.; Levi, M. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J. Biol. Chem. 2005, 280, 32317–32325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.X.; Jiang, T.; Shen, Y.; Adorini, L.; Pruzanski, M.; Gonzalez, F.J.; Scherzer, P.; Lewis, L.; Miyazaki-Anzai, S.; Levi, M. The farnesoid X receptor modulates renal lipid metabolism and diet-induced renal inflammation, fibrosis, and proteinuria. Am. J. Physiol. Renal Physiol. 2009, 297, F1587–F1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobulescu, I.A.; Dubree, M.; Zhang, J.; McLeroy, P.; Moe, O.W. Reduction of renal triglyceride accumulation: Effects on proximal tubule Na+/H+ exchange and urinary acidification. Am. J. Physiol. Renal Physiol. 2009, 297, F1419–F1426. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontes, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in kidney, heart, and skeletal muscle dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, R.J.; Pezzolesi, M.G.; Summers, S.A. Rotten to the cortex: Ceramide-mediated lipotoxicity in diabetic kidney disease. Front. Endocrinol. 2020, 11, 622692. [Google Scholar] [CrossRef]
- Escasany, E.; Izquierdo-Lahuerta, A.; Medina-Gomez, G. Underlying mechanisms of renal lipotoxicity in obesity. Nephron 2019, 143, 28–32. [Google Scholar] [CrossRef]
- Fassett, R.G.; Robertson, I.K.; Ball, M.J.; Geraghty, D.P.; Coombes, J.S. Effect of atorvastatin on kidney function in chronic kidney disease: A randomised double-blind placebo-controlled trial. Atherosclerosis 2010, 213, 218–224. [Google Scholar] [CrossRef]
- Haynes, R.; Lewis, D.; Emberson, J.; Reith, C.; Agodoa, L.; Cass, A.; Craig, J.C.; de Zeeuw, D.; Feldt-Rasmussen, B.; Fellstrom, B.; et al. Effects of lowering LDL cholesterol on progression of kidney disease. J. Am. Soc. Nephrol. 2014, 25, 1825–1833. [Google Scholar] [CrossRef] [Green Version]
- Vogt, L.; Bangalore, S.; Fayyad, R.; Melamed, S.; Hovingh, G.K.; DeMicco, D.A.; Waters, D.D. Atorvastatin has a dose-dependent beneficial effect on kidney function and associated cardiovascular outcomes: Post hoc analysis of 6 double-blind randomized controlled trials. J. Am. Heart Assoc. 2019, 8, e010827. [Google Scholar] [CrossRef] [Green Version]
- Esmeijer, K.; Dekkers, O.M.; de Fijter, J.W.; Dekker, F.W.; Hoogeveen, E.K. Effect of different types of statins on kidney function decline and proteinuria: A network meta-analysis. Sci. Rep. 2019, 9, 16632. [Google Scholar] [CrossRef]
- Gotoh, K.; Masaki, T.; Chiba, S.; Ando, H.; Fujiwara, K.; Shimasaki, T.; Tawara, Y.; Toyooka, I.; Shiraishi, K.; Mitsutomi, K.; et al. Effects of hydrophilic statins on renal tubular lipid accumulation in diet-induced obese mice. Obes. Res. Clin. Pract. 2013, 7, e342–e352. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Minami, S. Versuche an Überlebendem Carcinom-gewebe. Klin. Wochenschr. 1923, 2, 776–777. [Google Scholar] [CrossRef]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices In Vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.P.; de Cedron, M.G.; de Molina, A.R. Alterations of lipid metabolism in cancer: Implications in prognosis and treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef]
- Peck, B.; Schulze, A. Lipid metabolism at the nexus of diet and tumor microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J. Exp. Clin. Cancer Res. 2019, 38, 52. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Han, H.; Liu, L.; Duan, Y.; Yang, X.; Ma, C.; Zhu, Y.; Han, J.; Li, X.; Chen, Y. CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis 2018, 7, 98. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Thompson, B.; Han, S.; Lotan, Y.; McDonald, J.G.; Ye, J. Uptake of HDL-cholesterol contributes to lipid accumulation in clear cell renal cell carcinoma. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 158525. [Google Scholar] [CrossRef]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Wettersten, H.I.; Hakimi, A.A.; Morin, D.; Bianchi, C.; Johnstone, M.E.; Donohoe, D.R.; Trott, J.F.; Aboud, O.A.; Stirdivant, S.; Neri, B.; et al. Grade-dependent metabolic reprogramming in kidney cancer revealed by combined proteomics and metabolomics analysis. Cancer Res. 2015, 75, 2541–2552. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Y.; Liu, Q.; Wu, F.; Liu, X.; Qu, H.; Yuan, Y.; Ge, J.; Xu, Y.; Wang, H. The mRNA expression signature and prognostic analysis of multiple fatty acid metabolic enzymes in clear cell renal cell carcinoma. J. Cancer 2019, 10, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Arai, E.; Maekawa, K.; Ishikawa, M.; Fujimoto, H.; Taguchi, R.; Matsumoto, K.; Kanai, Y.; Saito, Y. Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Sci. Rep. 2016, 6, 28932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Ferro, M.; Loizzo, D.; Bianchi, C.; Terracciano, D.; Cantiello, F.; Bell, L.N.; Battaglia, S.; Porta, C.; Gernone, A.; et al. Integration of lipidomics and transcriptomics reveals reprogramming of the lipid metabolism and composition in clear cell renal cell carcinoma. Metabolites 2020, 10, 509. [Google Scholar] [CrossRef]
- Xu, G.H.; Lou, N.; Shi, H.C.; Xu, Y.C.; Ruan, H.L.; Xiao, W.; Liu, L.; Li, X.; Xiao, H.B.; Qiu, B.; et al. Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 88. [Google Scholar] [CrossRef]
- Riscal, R.; Bull, C.J.; Mesaros, C.; Finan, J.M.; Carens, M.; Ho, E.S.; Xu, J.P.; Godfrey, J.; Brennan, P.; Johansson, M.; et al. Cholesterol auxotrophy as a targetable vulnerability in clear cell renal cell carcinoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Chou, Y.C.; Lin, C.H.; Wong, C.S.; Chou, W.Y.; Chang, J.Y.; Sun, C.A. Statin use and the risk of renal cell carcinoma: National cohort study. J. Investig. Med. 2020, 68, 776–781. [Google Scholar] [CrossRef]
- Khurana, V.; Caldito, G.; Ankem, M. Statins might reduce risk of renal cell carcinoma in humans: Case-control study of 500,000 veterans. Urology 2008, 71, 118–122. [Google Scholar] [CrossRef]
- Goldstein, M.R.; Mascitelli, L.; Pezzetta, F.; Khurana, V. Statins might reduce risk of renal cell carcinoma in humans: Case-control study of 500,000 veterans (Urology 2008;71:118–122). Urology 2008, 72, 717. [Google Scholar] [CrossRef]
- Thompson, J.M.; Alvarez, A.; Singha, M.K.; Pavesic, M.W.; Nguyen, Q.H.; Nelson, L.J.; Fruman, D.A.; Razorenova, O.V. Targeting the mevalonate pathway suppresses VHL-deficient CC-RCC through an HIF-dependent mechanism. Mol. Cancer Ther. 2018, 17, 1781–1792. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Clark, P.E.; Cookson, M.S. The von Hippel-Lindau gene: Turning discovery into therapy. Cancer 2008, 113 (Suppl. S7), 1768–1778. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Shen, C.; Kaelin, W.G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2013, 23, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nargund, A.M.; Pham, C.G.; Dong, Y.; Wang, P.I.; Osmangeyoglu, H.U.; Xie, Y.; Aras, O.; Han, S.; Oyama, T.; Takeda, S.; et al. The SWI/SNF protein PBRM1 restrains VHL-loss-driven clear cell renal cell carcinoma. Cell Rep. 2017, 18, 2893–2906. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, W.; Xiao, T.; Liu, X.S.; Kaelin, W.G., Jr. Inactivation of the PBRM1 tumor suppressor gene amplifies the HIF-response in VHL-/- clear cell renal carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-inducible factors and the regulation of lipid metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, I.H.; Calvo, S.E.; Markhard, A.L.; Skinner, O.S.; To, T.L.; Ast, T.; Mootha, V.K. Genetic screen for cell fitness in high or low oxygen highlights mitochondrial and lipid metabolism. Cell 2020, 181, 716–727.e11. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyhan, M.J.; El Mashad, S.M.; O’Donovan, T.R.; Ahmad, S.; Collins, C.; Sweeney, P.; Rogers, E.; O’Sullivan, G.C.; McKenna, S.L. VHL genetic alteration in CCRCC does not determine de-regulation of HIF, CAIX, hnRNP A2/B1 and osteopontin. Anal. Cell Pathol. 2010, 33, 121–132. [Google Scholar] [CrossRef]
- Li, X.J.; Li, Q.L.; Ju, L.G.; Zhao, C.; Zhao, L.S.; Du, J.W.; Wang, Y.; Zheng, L.; Song, B.L.; Li, L.Y.; et al. Deficiency of histone methyltransferase SET domain-containing 2 in liver leads to abnormal lipid metabolism and HCC. Hepatology 2021, 73, 1797–1815. [Google Scholar] [CrossRef]
- Xu, W.H.; Qu, Y.Y.; Wang, J.; Wang, H.K.; Wan, F.N.; Zhao, J.Y.; Zhang, H.L.; Ye, D.W. Elevated CD36 expression correlates with increased visceral adipose tissue and predicts poor prognosis in ccRCC patients. J. Cancer 2019, 10, 4522–4531. [Google Scholar] [CrossRef]
- Kim, Y.S.; Jung, J.; Jeong, H.; Lee, J.H.; Oh, H.E.; Lee, E.S.; Choi, J.W. High membranous expression of fatty acid transport protein 4 is associated with tumorigenesis and tumor progression in clear cell renal cell carcinoma. Dis. Markers 2019, 2019, 5702026. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, Y.; Garbacz, W.G.; Jiang, M.; Xu, M.; Huang, H.; Tsung, A.; Billiar, T.R.; Ramakrishnan, S.K.; Shah, Y.M.; et al. Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J. Hepatol. 2015, 63, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W.; et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, D.J.; Simon, M.C. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2018, 1870, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Chow, W.H.; Gridley, G.; Fraumeni, J.F., Jr.; Jarvholm, B. Obesity, hypertension, and the risk of kidney cancer in men. N. Engl. J. Med. 2000, 343, 1305–1311. [Google Scholar] [CrossRef]
- Sanfilippo, K.M.; McTigue, K.M.; Fidler, C.J.; Neaton, J.D.; Chang, Y.; Fried, L.F.; Liu, S.; Kuller, L.H. Hypertension and obesity and the risk of kidney cancer in 2 large cohorts of US men and women. Hypertension 2014, 63, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Qin, B.; Poti, J.; Sokol, R.; Gordon-Larsen, P. Epidemiology of obesity in adults: Latest trends. Curr. Obes. Rep. 2018, 7, 276–288. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017–2018; NCHS Data Brief, no 360; National Center for Health Statistics: Hyattsville, MD, USA, 2020. [Google Scholar]
- Gan, C.L.; Heng, D.Y.C. New insights into the obesity paradox in renal cell carcinoma. Nat. Rev. Nephrol. 2020, 16, 253–254. [Google Scholar] [CrossRef]
- Donin, N.M.; Pantuck, A.; Klopfer, P.; Bevan, P.; Fall, B.; Said, J.; Belldegrun, A.S.; Chamie, K. Body mass index and survival in a prospective randomized trial of localized high-risk renal cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.H.; Doan, P.; He, Y.; Lau, H.M.; Pleass, H.; Patel, M.I. A systematic review and meta-analysis of the significance of body mass index on kidney cancer outcomes. J. Urol. 2021, 205, 346–355. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The obesity paradox in cancer: A review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Giovannucci, E.L. The obesity paradox in cancer: Epidemiologic insights and perspectives. Curr. Nutr. Rep. 2019, 8, 175–181. [Google Scholar] [CrossRef]
- Petrelli, F.; Cortellini, A.; Indini, A.; Tomasello, G.; Ghidini, M.; Nigro, O.; Salati, M.; Dottorini, L.; Iaculli, A.; Varricchio, A.; et al. Association of obesity with survival outcomes in patients with cancer: A systematic review and meta-analysis. JAMA Netw. Open 2021, 4, e213520. [Google Scholar] [CrossRef]
- Hainer, V.; Aldhoon-Hainerova, I. Obesity paradox does exist. Diabetes Care 2013, 36 (Suppl. S2), S276–S281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Bu, R. Biological support to obesity paradox in renal cell carcinoma: A review. Urol. Int. 2020, 104, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.S.; Lohse, C.M.; Cheville, J.C.; Thiel, D.D.; Leibovich, B.C.; Blute, M.L. Greater body mass index is associated with better pathologic features and improved outcome among patients treated surgically for clear cell renal cell carcinoma. Urology 2006, 68, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: A cohort study. Lancet Oncol. 2020, 21, 283–293. [Google Scholar] [CrossRef]
- Ito, R.; Narita, S.; Huang, M.; Nara, T.; Numakura, K.; Takayama, K.; Tsuruta, H.; Maeno, A.; Saito, M.; Inoue, T.; et al. The impact of obesity and adiponectin signaling in patients with renal cell carcinoma: A potential mechanism for the ”obesity paradox”. PLoS ONE 2017, 12, e0171615. [Google Scholar] [CrossRef] [PubMed]
- Turco, F.; Tucci, M.; Di Stefano, R.F.; Samuelly, A.; Bungaro, M.; Audisio, M.; Pisano, C.; Di Maio, M.; Scagliotti, G.V.; Buttigliero, C. Renal cell carcinoma (RCC): Fatter is better? A review on the role of obesity in RCC. Endocr. Relat. Cancer 2021, 28, R207–R216. [Google Scholar] [PubMed]
- Banack, H.R.; Stokes, A. The ’obesity paradox’ may not be a paradox at all. Int. J. Obes. 2017, 41, 1162–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. International agency for research on cancer handbook working, body fatness and cancer—Viewpoint of the IARC working group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Basen-Engquist, K.; Chang, M. Obesity and cancer risk: Recent review and evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, inflammation, and cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut microbiota and cancer: From pathogenesis to therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tu, H.; Zhu, M.; Liang, D.; Ye, Y.; Chang, D.W.; Long, Y.; Wu, X. Circulating obesity-driven biomarkers are associated with risk of clear cell renal cell carcinoma: A two-stage, case-control study. Carcinogenesis 2019, 40, 1191–1197. [Google Scholar] [CrossRef]
- Kaminska, K.; Czarnecka, A.M.; Escudier, B.; Lian, F.; Szczylik, C. Interleukin-6 as an emerging regulator of renal cell cancer. Urol. Oncol. 2015, 33, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.M.; Weinstein, S.J.; Pollak, M.; Li, Z.; Virtamo, J.; Albanes, D.; Chow, W.H.; Purdue, M.P. Prediagnostic circulating adipokine concentrations and risk of renal cell carcinoma in male smokers. Carcinogenesis 2013, 34, 109–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, L.M.; Schwartz, K.; Pollak, M.; Graubard, B.I.; Li, Z.; Ruterbusch, J.; Rothman, N.; Davis, F.; Wacholder, S.; Colt, J.; et al. Serum leptin and adiponectin levels and risk of renal cell carcinoma. Obesity 2013, 21, 1478–1485. [Google Scholar] [CrossRef] [Green Version]
- Liao, L.M.; Hofmann, J.N.; Cho, E.; Pollak, M.N.; Chow, W.H.; Purdue, M.P. Circulating levels of obesity-related markers and risk of renal cell carcinoma in the PLCO cancer screening trial. Cancer Causes Control 2017, 28, 801–807. [Google Scholar] [CrossRef]
- Dimou, N.L.; Papadimitriou, N.; Mariosa, D.; Johansson, M.; Brennan, P.; Peters, U.; Chanock, S.J.; Purdue, M.; Bishop, D.T.; Gago-Dominquez, M.; et al. Circulating adipokine concentrations and risk of five obesity-related cancers: A Mendelian randomization study. Int. J. Cancer 2021, 148, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Karagozian, R.; Derdak, Z.; Baffy, G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism 2014, 63, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Ganne-Carrie, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Wu, G.Z.; Goh, K.J.; Lee, Y.M.; Ng, C.C.; You, A.B.; Wang, J.; Jia, D.; Hao, A.; Yu, Q.; et al. Saturated fatty acids modulate cell response to DNA damage: Implication for their role in tumorigenesis. PLoS ONE 2008, 3, e2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef]
- Ge, M.; Fontanesi, F.; Merscher, S.; Fornoni, A. The vicious cycle of renal lipotoxicity and mitochondrial dysfunction. Front. Physiol. 2020, 11, 732. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [Green Version]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Bratslavsky, G.; Sudarshan, S.; Neckers, L.; Linehan, W.M. Pseudohypoxic pathways in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 4667–4671. [Google Scholar] [CrossRef] [Green Version]
- Saville, K.M.; Clark, J.; Wilk, A.; Rogers, G.D.; Andrews, J.F.; Koczor, C.A.; Sobol, R.W. NAD(+)-mediated regulation of mammalian base excision repair. DNA Repair 2020, 93, 102930. [Google Scholar] [CrossRef]
- Lair, B.; Laurens, C.; Van Den Bosch, B.; Moro, C. Novel insights and mechanisms of lipotoxicity-driven insulin resistance. Int. J. Mol. Sci. 2020, 21, 6358. [Google Scholar] [CrossRef]
- Azevedo-Martins, A.K.; Monteiro, A.P.; Lima, C.L.; Lenzen, S.; Curi, R. Fatty acid-induced toxicity and neutral lipid accumulation in insulin-producing RINm5F cells. Toxicol. In Vitro 2006, 20, 1106–1113. [Google Scholar] [CrossRef]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Inagi, R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp. Nephrol. 2009, 112, e1–e9. [Google Scholar] [CrossRef]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobulescu, I.A.; Pop, L.M.; Mani, C.; Turner, K.; Rivera, C.; Khatoon, S.; Kairamkonda, S.; Hannan, R.; Palle, K. Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma. Metabolites 2021, 11, 608. https://doi.org/10.3390/metabo11090608
Bobulescu IA, Pop LM, Mani C, Turner K, Rivera C, Khatoon S, Kairamkonda S, Hannan R, Palle K. Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma. Metabolites. 2021; 11(9):608. https://doi.org/10.3390/metabo11090608
Chicago/Turabian StyleBobulescu, Ion Alexandru, Laurentiu M. Pop, Chinnadurai Mani, Kala Turner, Christian Rivera, Sabiha Khatoon, Subash Kairamkonda, Raquibul Hannan, and Komaraiah Palle. 2021. "Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma" Metabolites 11, no. 9: 608. https://doi.org/10.3390/metabo11090608