
Altered Germinal-Center Metabolism in B Cells in Autoimmunity


Abstract
:
1. Introduction
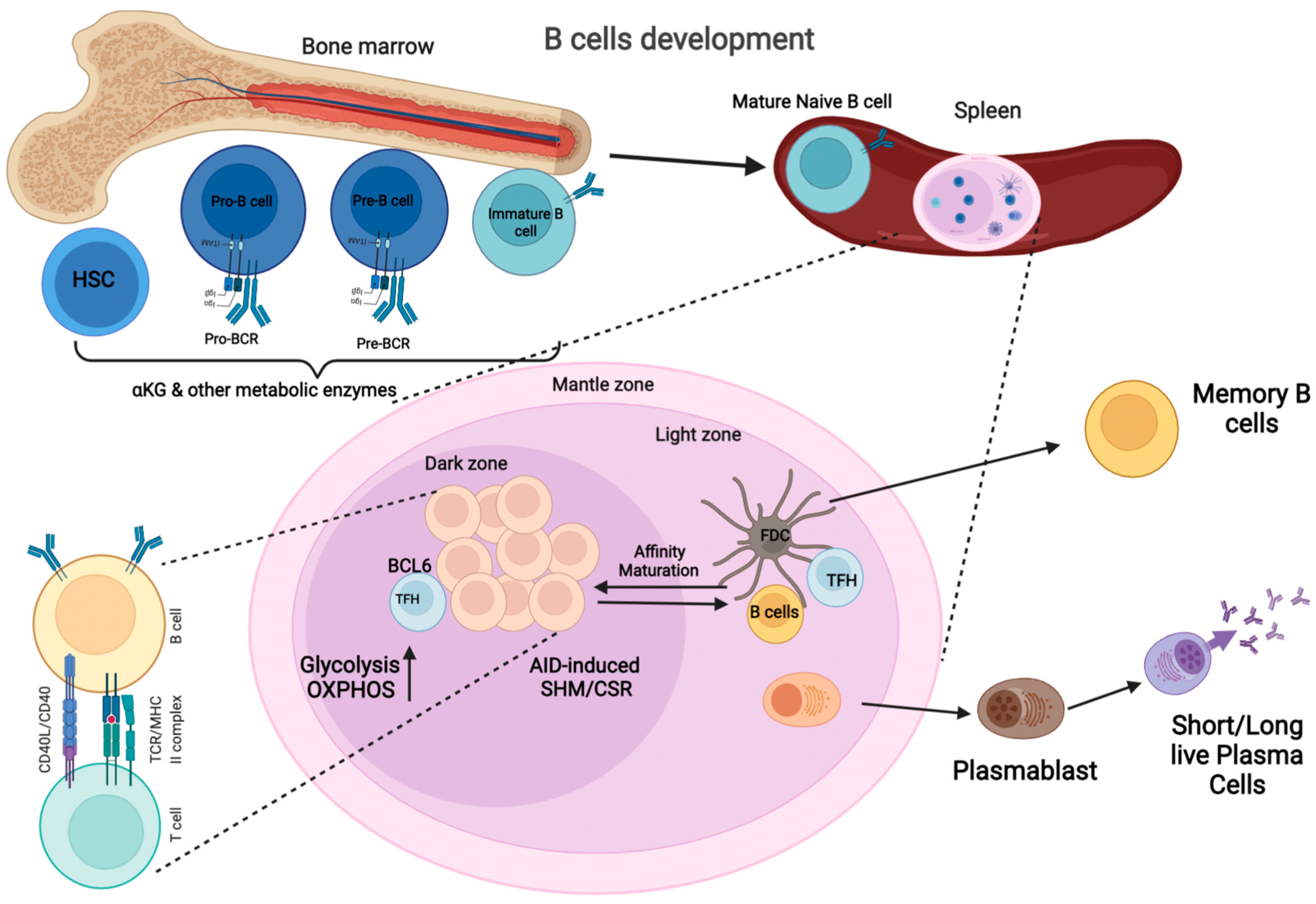
2. Germinal Center Reaction
3. B-Cell-Activating Factor and B-Cell Survival
3.1. Tolerogenicity in B-Cell Metabolism
3.2. Cytokine Signaling in B-Cell Metabolism
3.3. Oxidative Phosphorylation in B Cells
3.4. Lipid Metabolism in B Cells
3.5. Antioxidant in B-Cell Metabolism
3.6. B-Cell Differentiation and Plasma Cells
3.7. Amino Acids and B-Cell Metabolism
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hampe, C.S. B Cells in Autoimmune Diseases. Scientifica 2012, 2012, 215308. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.-H.; Jones, R.G. Fueling Immunity: Insights into Metabolism and Lymphocyte Function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.H.; Eisenberg, R.A. The role of toll-like receptors in systemic lupus erythematosus. Springer Semin. Immunopathol. 2006, 28, 131–143. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iperi, C.; Bordron, A.; Dueymes, M.; Pers, J.-O.; Jamin, C. Metabolic Program of Regulatory B Lymphocytes and Influence in the Control of Malignant and Autoimmune Situations. Front. Immunol. 2021, 12, 3984. [Google Scholar] [CrossRef] [PubMed]
- Raza, I.G.A.; Clarke, A.J. B Cell Metabolism and Autophagy in Autoimmunity. Front. Immunol. 2021, 12, 681105. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.S.; Moore, D.J. B Cell Metabolism: An Understudied Opportunity to Improve Immune Therapy in Autoimmune Type 1 Diabetes. Immunometabolism 2020, 2, 200016. [Google Scholar] [CrossRef] [Green Version]
- Khalsa, J.; Chawla, A.S.; Prabhu, S.B.; Vats, M.; Dhar, A.; Dev, G.; Das, N.; Mukherjee, S.; Tanwar, S.; Banerjee, H.; et al. Functionally significant metabolic differences between B and T lymphocyte lineages. Immunology 2019, 158, 104–120. [Google Scholar] [CrossRef]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.M.; Opipari, A.W., Jr.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisel, F.J.; Mullett, S.J.; Elsner, R.A.; Menk, A.V.; Trivedi, N.; Luo, W.; Wikenheiser, D.; Hawse, W.F.; Chikina, M.; Smita, S.; et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 2020, 21, 331–342. [Google Scholar] [CrossRef]
- MacIver, N.J.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Coloff, J.L.; Rathmell, J.C. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukoc. Biol. 2008, 84, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [Green Version]
- Oropallo, M.A.; Cerutti, A. Germinal center reaction: Antigen affinity and presentation explain it all. Trends Immunol. 2014, 35, 287–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Weisel, F.; Shlomchik, M.J. B Cell Receptor and CD40 Signaling Are Rewired for Synergistic Induction of the c-Myc Transcription Factor in Germinal Center B Cells. Immunity 2018, 48, 313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-C.; Morel, L. Immune metabolism regulation of the germinal center response. Exp. Mol. Med. 2020, 52, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Egawa, T.; Bhattacharya, D. Regulation of metabolic supply and demand during B cell activation and subsequent differentiation. Curr. Opin. Immunol. 2019, 57, 8–14. [Google Scholar] [CrossRef]
- Ersching, J.; Efeyan, A.; Mesin, L.; Jacobsen, J.T.; Pasqual, G.; Grabiner, B.C.; Dominguez-Sola, D.; Sabatini, D.M.; Victora, G.D. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity 2017, 46, 1045–1058.e6. [Google Scholar] [CrossRef] [Green Version]
- Omori, S.A.; Rickert, R.C. Phosphatidylinositol 3-kinase (PI3K) signaling and regulation of the antibody response. Cell Cycle 2007, 6, 397–402. [Google Scholar] [CrossRef]
- Mendoza, P.; Martínez-Martín, N.; Bovolenta, E.R.; Reyes-Garau, D.; Hernansanz-Agustín, P.; Delgado, P.; Diaz-Muñoz, M.D.; Oeste, C.L.; Fernández-Pisonero, I.; Castellano, E.; et al. R-Ras2 is required for germinal center formation to aid B cells during energetically demanding processes. Sci. Signal. 2018, 11, eaal1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.H.; Raybuck, A.L.; Stengel, K.; Wei, M.; Beck, T.C.; Volanakis, E.; Thomas, J.W.; Hiebert, S.; Haase, V.H.; Boothby, M.R. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 2016, 537, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellusova, J.; Cato, M.H.; Apgar, J.R.; Ramezani-Rad, P.; Leung, C.R.; Chen, C.; Richardson, A.D.; Conner, E.M.; Benschop, E.M.C.R.J.; Woodgett, J.R.; et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017, 18, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. 2018, 9, 129. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Ohteki, T.; Parsons, M.; Zakarian, A.; Jones, R.G.; Nguyen, L.T.; Woodgett, J.; Ohashi, P.S. Negative Regulation of T Cell Proliferation and Interleukin 2 Production by the Serine Threonine Kinase Gsk-3. J. Exp. Med. 2000, 192, 99–104. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3560. [Google Scholar] [CrossRef] [Green Version]
- Olson, A.L.; Pessin, J.E. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr. 1996, 16, 235–256. [Google Scholar] [CrossRef]
- Jayachandran, N.; Mejia, E.M.; Sheikholeslami, K.; Sher, A.A.; Hou, S.; Hatch, G.M.; Marshall, A.J. TAPP Adaptors Control B Cell Metabolism by Modulating the Phosphatidylinositol 3-Kinase Signaling Pathway: A Novel Regulatory Circuit Preventing Autoimmunity. J. Immunol. 2018, 201, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.; Hanczko, R.; Doherty, E. Assessment of mitochondrial dysfunction in lymphocytes of patients with systemic lupus erythematosus. Methods Mol. Biol. 2012, 900, 61–89. [Google Scholar] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic but Exaggerated in Chronically BAFF-Exposed B Cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor–mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Blair, D.; Dufort, F.J.; Chiles, T.C. Protein kinase Cbeta is critical for the metabolic switch to glycolysis following B-cell antigen receptor engagement. Biochem. J. 2012, 448, 165–169. [Google Scholar] [CrossRef]
- Tsui, C.; Martinez-Martin, N.; Gaya, M.; Maldonado, P.; Llorian, M.; Legrave, N.M.; Rossi, M.; MacRae, J.I.; Cameron, A.J.; Parker, P.J.; et al. Protein Kinase C-beta Dictates B Cell Fate by Regulating Mitochondrial Remodeling, Metabolic Reprogramming, and Heme Biosynthesis. Immunity 2018, 48, 1144–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Salmond, R.J. mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Morel, L.; Croker, B.P.; Blenman, K.R.; Mohan, C.; Huang, G.; Gilkeson, G.; Wakeland, E.K. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc. Natl. Acad. Sci. USA 2000, 97, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Hawse, W.; Conter, L.; Trivedi, N.; Weisel, F.; Wikenheiser, D.; Cattley, R.T.; Shlomchik, M.J. The AKT kinase signaling network is rewired by PTEN to control proximal BCR signaling in germinal center B cells. Nat. Immunol. 2019, 20, 736–746. [Google Scholar] [CrossRef]
- Okada, T.; Moriyama, S.; Kitano, M. Differentiation of germinal center B cells and follicular helper T cells as viewed by tracking Bcl6 expression dynamics. Immunol. Rev. 2012, 247, 120–132. [Google Scholar] [CrossRef]
- Finkin, S.; Hartweger, H.; Oliveira, T.Y.; Kara, E.E.; Nussenzweig, M.C. Protein Amounts of the MYC Transcription Factor Determine Germinal Center B Cell Division Capacity. Immunity 2019, 51, 324–336.e5. [Google Scholar] [CrossRef] [PubMed]
- Haniuda, K.; Fukao, S.; Kitamura, D. Metabolic Reprogramming Induces Germinal Center B Cell Differentiation through Bcl6 Locus Remodeling. Cell Rep. 2020, 33, 108333. [Google Scholar] [CrossRef]
- Teng, X.; Brown, J.; Choi, S.; Li, W.; Morel, L. Metabolic determinants of lupus pathogenesis. Immunol. Rev. 2020, 295, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-C.; Titov, A.A.; Abboud, G.; Seay, H.R.; Brusko, T.M.; Roopenian, D.C.; Salek-Ardakani, S.; Morel, L. Inhibition of glucose metabolism selectively targets autoreactive follicular helper T cells. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Choi, S.-C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abboud, G.; Choi, S.-C.; Kanda, N.; Zeumer-Spataro, L.; Roopenian, D.C.; Morel, L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1973. [Google Scholar] [CrossRef]
- Yin, Y.; Choi, S.C.; Xu, Z.; Zeumer, L.; Kanda, N.; Croker, B.P.; Morel, L. Glucose Oxidation Is Critical for CD4+ T Cell Activation in a Mouse Model of Systemic Lupus Erythematosus. J. Immunol. 2016, 196, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, N.M.; Marshak-Rothstein, A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin. Immunol. 2011, 23, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Suthers, A.N.; Sarantopoulos, S. TLR7/TLR9- and B Cell Receptor-Signaling Crosstalk: Promotion of Potentially Dangerous B Cells. Front. Immunol. 2017, 8, 775. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, C.; Celeghini, C.; Monasta, L.; Zauli, G. NF-κB pathways in hematological malignancies. Cell. Mol. Life Sci. 2014, 71, 2083–2102. [Google Scholar] [CrossRef]
- Ganeshan, K.; Chawla, A. Metabolic Regulation of Immune Responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [Green Version]
- Basso, K.; Dalla-Favera, R. BCL6: Master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol. 2010, 105, 193–210. [Google Scholar]
- Smulski, C.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 Kinase Signals BCR-Dependent Mature B Cell Survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Patke, A.; Mecklenbräuker, I.; Erdjument-Bromage, H.; Tempst, P.; Tarakhovsky, A. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J. Exp. Med. 2006, 203, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-X.; Wang, H.-Y.; Yin, L.; Mao, Y.-Y.; Zhou, W. Immunometabolism in the pathogenesis of systemic lupus erythematosus. J. Transl. Autoimmun. 2020, 3, 100046. [Google Scholar] [CrossRef] [PubMed]
- Moisini, I.; Davidson, A. BAFF: A local and systemic target in autoimmune diseases. Clin. Exp. Immunol. 2009, 158, 155–163. [Google Scholar] [CrossRef]
- Thien, M.; Phan, T.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Boneparth, A.; Davidson, A. B-cell activating factor targeted therapy and lupus. Arthritis Res. Ther. 2012, 14, S2. [Google Scholar] [CrossRef] [Green Version]
- Cancro, M.P. The BLyS/BAFF family of ligands and receptors: Key targets in the therapy and understanding of autoimmunity. Ann. Rheum. Dis. 2006, 65, iii34–iii36. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, H.; Qin, J.; Xu, Z.; Gui, L.; Liu, B.; Liu, C.; Xu, C.; Liu, W.; Zhang, S.; et al. Rapamycin inhibits BAFF-stimulated cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells. Cell Mol. Life Sci. 2015, 72, 4867–4884. [Google Scholar] [CrossRef]
- Wu, T.; Qin, X.; Kurepa, Z.; Kumar, K.R.; Liu, K.; Kanta, H.; Zhou, X.J.; Satterthwaite, A.B.; Davis, L.S.; Mohan, C. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J. Clin. Investig. 2007, 117, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulou, C.; Nikoleri, D.; Bertsias, G. Immunometabolism: An overview and therapeutic prospects in autoimmune diseases. Immunotherapy 2019, 11, 813–829. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookens, S.K.; Cho, S.H.; Basso, P.J.; Boothby, M.R. AMPKalpha1 in B Cells Dampens Primary Antibody Responses yet Promotes Mitochondrial Homeostasis and Persistence of B Cell Memory. J. Immunol. 2020, 205, 3011–3022. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Moon, S.J.; Kim, E.K.; Seo, H.B.; Yang, E.J.; Son, H.J.; Kim, J.K.; Min, J.K.; Park, S.H.; Cho, M.L. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J. Immunol. 2017, 198, 2661–2670. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Yang, T.; Liu, G.; Liu, H.; Peng, Y.; He, L. Piperine ameliorated lupus nephritis by targeting AMPK-mediated activation of NLRP3 inflammasome. Int. Immunopharmacol. 2018, 65, 448–457. [Google Scholar] [CrossRef]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Akkaya, M.; Traba, J.; Roesler, A.; Miozzo, P.; Akkaya, B.; Theall, B.P.; Sohn, H.; Pena, M.; Smelkinson, M.; Kabat, J.; et al. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol. 2018, 19, 871–884. [Google Scholar] [CrossRef]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, J.; Ng, Y.-S.; Coupillaud, C.; Paget, D.; Meffre, E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J. Exp. Med. 2005, 201, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P.; Grossman, C.; Niland, B.; Puskas, F.; Neupane, H.; Allam, F.; Banki, K.; Phillips, P.E.; Perl, A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 175–190. [Google Scholar] [CrossRef]
- Becker, A.M.; Dao, K.H.; Han, B.K.; Kornu, R.; Lakhanpal, S.; Mobley, A.B.; Li, Q.-Z.; Lian, Y.; Wu, T.; Reimold, A.M.; et al. SLE Peripheral Blood B Cell, T Cell and Myeloid Cell Transcriptomes Display Unique Profiles and Each Subset Contributes to the Interferon Signature. PLoS ONE 2013, 8, e67003. [Google Scholar] [CrossRef]
- Wu, B.; Goronzy, J.J.; Weyand, C.M. Metabolic Fitness of T Cells in Autoimmune Disease. Immunometabolism 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Sumikawa, M.H.; Iwata, S.; Zhang, M.; Miyata, H.; Ueno, M.; Todoroki, Y.; Nagayasu, A.; Kanda, R.; Sonomoto, K.; Torimoto, K.; et al. An enhanced mitochondrial function through glutamine metabolism in plasmablast differentiation in systemic lupus erythematosus. Rheumatology 2021, keab824. [Google Scholar] [CrossRef]
- Dufort, F.J.; Bleiman, B.F.; Gumina, M.R.; Blair, D.; Wagner, D.J.; Roberts, M.F.; Abu-Amer, Y.; Chiles, T.C. Cutting Edge: IL-4-Mediated Protection of Primary B Lymphocytes from Apoptosis via Stat6-Dependent Regulation of Glycolytic Metabolism. J. Immunol. 2007, 179, 4953–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guleria, A.; Pratap, A.; Dubey, D.; Rawat, A.; Chaurasia, S.; Sukesh, E.; Phatak, S.; Ajmani, S.; Kumar, U.; Khetrapal, C.L.; et al. NMR based serum metabolomics reveals a distinctive signature in patients with Lupus Nephritis. Sci. Rep. 2016, 6, 35309. [Google Scholar] [CrossRef] [Green Version]
- Nicolaou, O.; Kousios, A.; Hadjisavvas, A.; Lauwerys, B.; Sokratous, K.; Kyriacou, K. Biomarkers of systemic lupus erythematosus identified using mass spectrometry-based proteomics: A systematic review. J. Cell. Mol. Med. 2016, 21, 993–1012. [Google Scholar] [CrossRef]
- Brookens, S.K.; Boothby, M.R. AMPK Metabolism in the B Lineage Modulates Humoral Responses. Immunometabolism 2021, 3, e210011. [Google Scholar] [CrossRef]
- Lee, S.U.; Maeda, T. POK/ZBTB proteins: An emerging family of proteins that regulate lymphoid development and function. Immunol. Rev. 2012, 247, 107–119. [Google Scholar] [CrossRef]
- Doherty, E.; Oaks, Z.; Perl, A. Increased Mitochondrial Electron Transport Chain Activity at Complex I Is Regulated by N-Acetylcysteine in Lymphocytes of Patients with Systemic Lupus Erythematosus. Antioxid. Redox Signal. 2014, 21, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nieuwenhove, E.; Garcia-Perez, J.E.; Helsen, C.; Rodriguez, P.D.; van Schouwenburg, P.A.; Dooley, J.; Schlenner, S.; van der Burg, M.; Verhoeyen, E.; Gijsbers, R.; et al. A kindred with mutant IKAROS and autoimmunity. J. Allergy Clin. Immunol. 2018, 142, 699–702.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.; Xiao, G.; Geng, H.; Cosgun, K.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nat. Cell Biol. 2017, 542, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Chan, L.N.; Klemm, L.; Braas, D.; Chen, Z.; Geng, H.; Zhang, Q.C.; Aghajanirefah, A.; Cosgun, K.; Sadras, T.; et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell 2018, 173, 470–484.e18. [Google Scholar] [CrossRef]
- Huss, J.M.; Garbacz, W.G.; Xie, W. Constitutive activities of estrogen-related receptors: Transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim. Biophys. Acta 2015, 1852, 1912–1927. [Google Scholar] [CrossRef] [Green Version]
- Perry, D.J.; Yin, Y.; Telarico, T.; Baker, H.V.; Dozmorov, I.; Perl, A.; Morel, L. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor gamma. J. Immunol. 2012, 189, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Rathmell, J.; Pearce, E. SnapShot: Immunometabolism. Cell Metab. 2015, 22, 190–190.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightfoot, Y.L.; Blanco, L.P.; Kaplan, M.J. Metabolic abnormalities and oxidative stress in lupus. Curr. Opin. Rheumatol. 2017, 29, 442–449. [Google Scholar] [CrossRef]
- Price, M.; Patterson, D.; Scharer, C.; Boss, J.M. Progressive Upregulation of Oxidative Metabolism Facilitates Plasmablast Differentiation to a T-Independent Antigen. Cell Rep. 2018, 23, 3152–3159. [Google Scholar] [CrossRef]
- Schweighoffer, E.; Nys, J.; Vanes, L.; Smithers, N.; Tybulewicz, V.L. TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK. J. Exp. Med. 2017, 214, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, A.A.; Ross, R.; Fitzgerald, G.F.; Caplice, N.; Stanton, C. Role of the Gut in Modulating Lipoprotein Metabolism. Curr. Cardiol. Rep. 2014, 16, 515. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Mitchell, J.; Delaney, N.; Tsokos, G.C. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J. Immunol. 2004, 172, 7821–7831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.P.; Oeser, A.; Solus, J.; Avalos, I.; Gebretsadik, T.; Shintani, A.; Linton, M.F.; Fazio, S.; Stein, C.M. Inflammatory mechanisms affecting the lipid profile in patients with systemic lupus erythematosus. J. Rheumatol. 2007, 34, 1849–1854. [Google Scholar] [PubMed]
- Kobayashi, A.; Ito, A.; Shirakawa, I.; Tamura, A.; Tomono, S.; Shindou, H.; Hedde, P.N.; Tanaka, M.; Tsuboi, N.; Ishimoto, T.; et al. Dietary Supplementation With Eicosapentaenoic Acid Inhibits Plasma Cell Differentiation and Attenuates Lupus Autoimmunity. Front. Immunol. 2021, 12, 650856. [Google Scholar] [CrossRef] [PubMed]
- Hanna Kazazian, N.; Wang, Y.; Roussel-Queval, A.; Marcadet, L.; Chasson, L.; Laprie, C.; Desnues, B.; Charaix, J.; Irla, M.; Alexopoulou, L. Lupus Autoimmunity and Metabolic Parameters Are Exacerbated Upon High Fat Diet-Induced Obesity Due to TLR7 Signaling. Front. Immunol. 2019, 10, 2015. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.C.; Frieda, S.; Pearce, A.; Silverstein, R.L. Oxidized LDL Binds to CD36 on Human Monocyte-Derived Macrophages and Transfected Cell Lines. Arter. Thromb. Vasc. Biol. 1995, 15, 269–275. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Won, W.-J.; Bachmann, M.F.; Kearney, J.F. CD36 Is Differentially Expressed on B Cell Subsets during Development and in Responses to Antigen. J. Immunol. 2007, 180, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Wan, D.W.; Anwar, K.; Merrill, J.T.; Wirkowski, P.A.; Shah, N.; Cronstein, B.N.; Chan, E.S.; Carsons, S.E. Enhanced CD36 scavenger receptor expression in THP-1 human monocytes in the presence of lupus plasma: Linking autoimmunity and atherosclerosis. Exp. Biol. Med. 2009, 234, 354–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, H.; Kodali, S.; Wang, J. Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion 2018, 41, 58–65. [Google Scholar] [CrossRef]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and Autoimmunity: A “New Deal” of an Old Drug. Front Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Mahajan, N.; Sah, S.; Nath, S.K.; Paudyal, B. Oxidative stress and its biomarkers in systemic lupus erythematosus. J. Biomed. Sci. 2014, 21, 23. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.M. Autoantibodies in pathology and cell biology. Cell 1991, 67, 841–842. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Lu, L. B Cell-Mediated Autoimmune Diseases. Adv. Exp. Med. Biol. 2020, 1254, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.J.; Metzler, G.; Wray-Dutra, M.; Jackson, S.W. Altered B cell signalling in autoimmunity. Nat. Rev. Immunol. 2017, 17, 421–436. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nat. Cell Biol. 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Muntjewerff, E.M.; Meesters, L.D.; Bogaart, G.V.D.; Revelo, N.H. Reverse Signaling by MHC-I Molecules in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 605958. [Google Scholar] [CrossRef] [PubMed]
- Yarosz, E.; Chang, C.-H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Lao, Y.; Teng, X.-L.; Li, S.; Zhou, Y.; Wang, F.; Guo, X.; Deng, S.; Chang, Y.; Wu, X.; et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.; Kiran, R.; Wanchu, A.; Bhatnagar, A. Soluble granzyme B and cytotoxic T lymphocyte activity in the pathogenesis of systemic lupus erythematosus. Cell. Immunol. 2011, 269, 16–21. [Google Scholar] [CrossRef]
- Ji, H.; Korganow, A.-S.; Mangialaio, S.; Hoglund, P.; Andre, I.; Luhder, F.; Gonzalez, A.; Poirot, L.; Benoist, C.; Mathis, D. Different modes of pathogenesis in T-cell-dependent autoimmunity: Clues from two TCR transgenic systems. Immunol. Rev. 1999, 169, 139–146. [Google Scholar] [CrossRef]
- Lam, W.Y.; Jash, A.; Yao, C.; D’Souza, L.; Wong, R.; Nunley, R.M.; Meares, G.P.; Patti, G.J.; Bhattacharya, D. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep. 2018, 24, 2479–2492.e6. [Google Scholar] [CrossRef] [Green Version]
- Lam, W.Y.; Becker, A.M.; Kennerly, K.M.; Wong, R.; Curtis, J.D.; Llufrio, E.M.; McCommis, K.; Fahrmann, J.; Pizzato, H.A.; Nunley, R.M.; et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immun. 2016, 45, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Kunisawa, J.; Sugiura, Y.; Wake, T.; Nagatake, T.; Suzuki, H.; Nagasawa, R.; Shikata, S.; Honda, K.; Hashimoto, E.; Suzuki, Y.; et al. Mode of Bioenergetic Metabolism during B Cell Differentiation in the Intestine Determines the Distinct Requirement for Vitamin B 1. Cell Rep. 2015, 13, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.; Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, Downstream of Blimp-1, Expands the Secretory Apparatus and Other Organelles, and Increases Protein Synthesis in Plasma Cell Differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, M.; Yim, S.H.; Garcia-Manteiga, J.M.; Masciarelli, S.; Kim, Y.-J.; Kang, M.-H.; Iuchi, Y.; Fujii, J.; Venè, R.; Rubartelli, A.; et al. B- to Plasma-Cell Terminal Differentiation Entails Oxidative Stress and Profound Reshaping of the Antioxidant Responses. Antioxid. Redox Signal. 2010, 13, 1133–1144. [Google Scholar] [CrossRef]
- Gass, J.N.; Gunn, K.E.; Sriburi, R.; Brewer, J.W. Stressed-out B cells? Plasma-cell differentiation and the unfolded protein response. Trends Immunol. 2004, 25, 17–24. [Google Scholar] [CrossRef]
- Sharabi, A.; Tsokos, G.C. T cell metabolism: New insights in systemic lupus erythematosus pathogenesis and therapy. Nat. Rev. Rheumatol. 2020, 16, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Yoshida, N.; Tsokos, G.C. Metabolic control of T cells in autoimmunity. Curr. Opin. Rheumatol. 2020, 32, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Cruzat, V.; Macedo Rogero, M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.D.; Gaudette, B.T.; Wilmore, J.R.; Chernova, I.; Bortnick, A.; Weiss, B.M.; Allman, D. mTOR has distinct functions in generating versus sustaining humoral immunity. J. Clin. Investig. 2016, 126, 4250–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosi, A.; Banfi, D.; Bistoletti, M.; Giaroni, C.; Baj, A. Tryptophan Metabolites Along the Microbiota-Gut-Brain Axis: An Interkingdom Communication System Influencing the Gut in Health and Disease. Int. J. Tryptophan Res. 2020, 13, 1178646920928984. [Google Scholar] [CrossRef]
- Widner, B.; Sepp, N.; Kowald, E.; Ortner, U.; Wirleitner, B.; Fritsch, P.; Baier-Bitterlich, G.; Fuchs, D. Enhanced Tryptophan Degradation in Systemic Lupus Erythematosus. Immunobiology 2000, 201, 621–630. [Google Scholar] [CrossRef]
- Fagone, P.; Sriburi, R.; Ward-Chapman, C.; Frank, M.; Wang, J.; Gunter, C.; Brewer, J.W.; Jackowski, S. Phospholipid Biosynthesis Program Underlying Membrane Expansion during B-lymphocyte Differentiation. J. Biol. Chem. 2007, 282, 7591–7605. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Altered Immunometabolism | Significance | Potential Treatment | |
|---|---|---|---|
| Enhanced lipid uptake due to the upregulation of CD36/fatty acid transport protein | Activation of inflammasomes | Etomoxir CD36 blocker | A |
| Enhanced glucose uptake | Enhanced glycolysis in naive B cells and altered glycosylation and function on immunoglobulin | 2DG or Glut1 inhibitors | B |
| Increase mitochondrial mass and function | Increased inhibition of regulatory phosphatase due to mitochondrial ROS | Metformin | C |
| ROS resistance | Resistance to metabolic stress | ROS inhibitor | D |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiraz, A.K.; Panther, E.J.; Reilly, C.M. Altered Germinal-Center Metabolism in B Cells in Autoimmunity. Metabolites 2022, 12, 40. https://doi.org/10.3390/metabo12010040
Shiraz AK, Panther EJ, Reilly CM. Altered Germinal-Center Metabolism in B Cells in Autoimmunity. Metabolites. 2022; 12(1):40. https://doi.org/10.3390/metabo12010040
Chicago/Turabian StyleShiraz, Ashton K., Eric J. Panther, and Christopher M. Reilly. 2022. "Altered Germinal-Center Metabolism in B Cells in Autoimmunity" Metabolites 12, no. 1: 40. https://doi.org/10.3390/metabo12010040