3.2. In Silico Results
In the current work, we investigated, using in silico molecular docking, the potential of the major extract’s components to interfere with three target proteins with known roles in mediating oxidative stress and the consequent inflammatory response, namely xanthine oxidase, heat shock protein 90, and inducible nitric oxide synthase.
The molecular docking served as a tool to explore which constituents of the extract are expected to demonstrate good fitting, and thus good binding affinity, towards the binding sites of the molecular targets. The binding affinity of the docked compound was estimated by the docking score parameter, which quantifies the stability of the binding between the docked pose and the amino acids lining the binding site. The top poses are further explored to confirm their interaction with crucial reported amino acids. For a top scoring compound, the mere presence of interactions with the crucial amino acids in the binding site does not suffice; rather, a top hit candidate must show minimum docking score relative to the reference drugs and the other compounds in the extract. Scoring stems from enthalpy and entropy changes, and it depends on good electronic complementarity as well as good steric complementarity and the 3D orientation of interacting partners plays a role in mediating interactions.
Xanthine oxidase is an enzyme that mediates the generation of uric acid via the oxidation of hypoxanthine to xanthine that is further oxidized by the enzyme to uric acid. The enzyme is classified among the oxidoreductases owing to its ability to produce some reactive oxygen species, such as superoxide and hydrogen peroxide. The regulation of the enzyme’s activity via xanthine oxidase inhibitors is expected to treat hyperuricemia, decrease oxidative stress, and attenuate the consequent pathophysiological inflammatory responses. The crystal structure of bovine xanthine oxidase complexed with the natural flavonoid inhibitor quercetin was explored (PDB ID: 3NVY). It is worth mentioning that the bovine form of the enzyme shares 90% sequence similarity to the human xanthine oxidase and the amino acids involved in the binding are strictly conserved [
24]. Polar groups of quercetin form hydrogen bonds to amino acids Glu802, Arg880, and Thr1010. The two fused rings of quercetin are sandwiched between the phenylalanine residues Phe914 and Phe1009. This resembles the binding modes of most of the substrates of xanthine oxidase. Additional van der Waals interactions are reported with the hydrophobic residues Leu873, Leu1014, Leu648, Val1011, Phe1013 [
24].
The docked compounds revealed docking scores on xanthine oxidase that spanned a range of −4.18 to −8.30 kcal/mol (
Table 2). Among these compounds, three docked candidates exhibited better binding scores than the docked co-crystallized ligand quercetin and the reference inhibitor febuxostat. These three compounds are 23, 31, and 33, which displayed docking scores of −8.30, −7.70, and −7.61 kcal/mol, respectively, in comparison to −5.81 and −7.55 kcal/mol for quercetin and febuxostat. Examining the docked poses, quercetin was found to preserve hydrogen bonding to the residue Glu802 as well as the hydrophobic interaction with Val1011. Docked febuxostat displayed polar interactions of the carboxylate moiety with Arg880 and Thr1010. The thiazole ring of febuxostat interacts via hydrogen bonding to Glu802 and Thr1010 and also via pi–pi stacking to the residue Phe914.
Heat shock proteins are closely related to oxidative stress and their overexpression during this state mediates inflammatory and immunomodulatory responses.
Heat shock protein 90 (Hsp90) is a homodimer of which each polypeptide chain consists of three regions: N-terminal ATP-binding domain, a middle domain, and a C-terminal domain. It is considered an attractive target due to its control of the cellular activity of several regulatory and signal transduction proteins [
25]. The crystal structure of the N-domain of Hsp90 was determined by X-ray crystallography complexed to the inhibitor FJ5 and deposited in the protein databank (ID: 5XRB). The inhibitor FJ5 and well-known natural product inhibitor radicicol were both docked simultaneously with the test compounds, serving as co-crystallized ligand and reference ligand, respectively. Reported interactions of several inhibitors from different classes to the ATP binding site of this protein include interactions with the highly conserved residue Asp93, as well as interactions with Leu48, Asn51, Asp93, Gly97, and Asp102 [
26,
27].
The docked compounds displayed binding modes within the ATP domain that reproduced the reported interactions, their docking scores range between −4.05 and −9.81 kcal/mol. Meanwhile, docking the inhibitors FJ5 and radicicol into the protein’s binding site affords docking scores of −8.27 and −6.77 kcal/mol, respectively. The co-crystallized ligand FJ5 validated the docking protocol since it exhibited a similar binding mode to the co-crystallized mode preserving the interactions with Ser52, Asp93, and Thr184. The reference inhibitor radicicol also displayed a hydrogen bond interaction with the amino acid Asp93. Seven out of the thirty-six docked test compounds (7, 15, 22, 23, 32, 38, and 39) exhibited better docking scores than both the co-crystallized and reference ligands. Docking results on Hsp90 are summarized in
Table 3.
A high level of ROS during oxidative stress induces the enzyme inducible nitric oxide synthase (iNOS) and thus increases the production of NO, which in conjunction with other ROS contributes greatly to oxidative stress. The enzyme is a NOS isoform expressed due to pro-inflammatory factors. Overproduction of iNOS leads to high NO levels, and is correlated with various diseases, such as sepsis and altered immune response. Consequently, the development of iNOS inhibitors is desirable since they are associated with the alleviation of various pathologies, such as diabetes and neurologic and respiratory diseases [
28]. Murine iNOS was reported to show 89% sequence similarity to human iNOS and amino acids involved in the binding are highly conserved [
29]. The enzyme was deposited in the protein databank complexed with an imidazo[4,5-b]pyridine derivative (MPW), PDB ID: 3NW2. The ligand accommodates a hydrophobic pocket formed of Val346 and Phe363 and displays a salt bridge with the polar residue Glu371. Another hydrogen bond is displayed with the residue Arg382. Moreover, hydrophobic contacts with Trp84, Met114, Ser256, Arg260 have been implicated by other iNOS inhibitors [
29].
In this study, MPW was re-docked into the iNOS active site. Additionally, the reported selective inhibitor
N-(3-(aminomethyl)benzyl)acetamidine (1400 W) was chosen to be docked simultaneously with our test compounds [
30]. The docked MPW adopts a conformation in the active site in which its aromatic rings display hydrophobic interactions with Ile195 and Trp366. Meanwhile, the refence inhibitor 1400 W shows hydrogen bonding to amino acids Asn364 and Glu371. The docked natural compounds demonstrated good affinities to the iNOS enzyme which was revealed by the docking scores ranging between −3.83 and −11.05 kcal/mol, of which 14 compounds displayed better scores than both co-crystallized and reference ligands. The docking results on iNOS are summarized in
Table 4.
In an attempt to further validate our docking procedure, all compounds achieving better scores than both the co-crystallized and reference inhibitors on each of the three proteins were further re-docked twice and average scores of the three docking trials were calculated. Simultaneously, the co-crystallized and reference substrates for each protein were re-docked. The results of the docking trials are found in
Table 5.
From the three docking trials, the best two ligands for each protein were explored. Compounds
23, 31 were the top scorers on xanthine oxidase with average docking scores of −8.30 and −7.65 kcal/mol, respectively. The average scores of quercetin and febuxostat were −5.82 and −7.54 kcal/mol, respectively. The interactions afforded by the docked pose of the highest scoring candidate
23 were investigated. The compound established hydrogen bonds to the residues Ser876 and Thr1010, arene–hydrogen interaction with Leu648, and pi–pi stacking with Phe914. Compound
23 appears in an orientation sandwiched between the two aromatic residues Phe914 and Phe1009 at the binding site of xanthine oxidase, similar to the binding mode of the co-crystallized substrate, quercetin. This demonstrates the stability of this ligand–enzyme complex (
Figure 2).
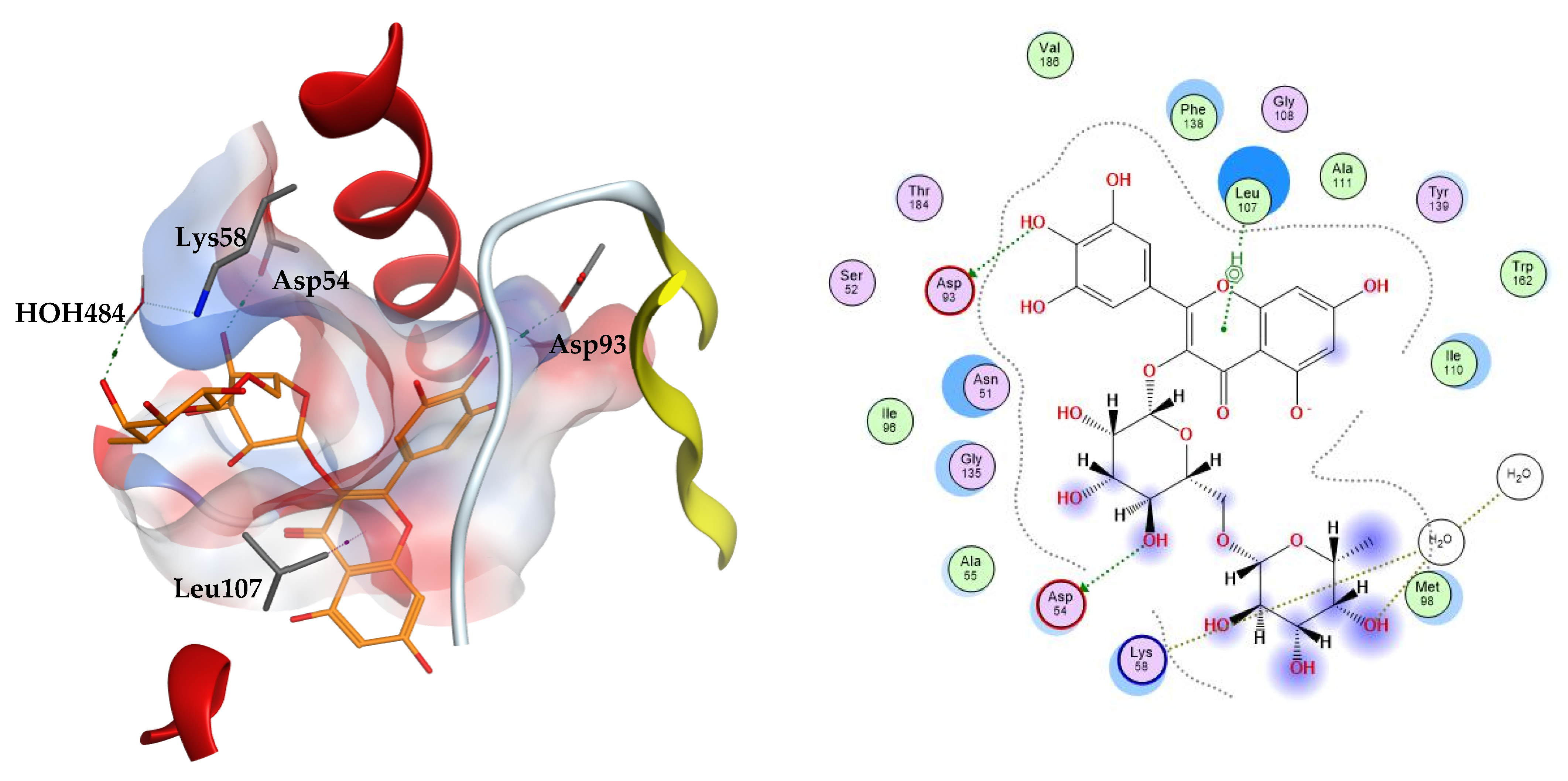
The two top scoring ligands on Hsp90 were compounds
32 and
39 with average docking scores of −9.92 and −9.64 kcal/mol, respectively. The average scores of docked FJ5 and radicicol were −8.47 and −6.16 kcal/mol, respectively. The best scoring candidate, compound
32, displayed arene–hydrogen interaction with Leu107, direct hydrogen bonding to Asp54 and Asp93, in addition to indirect hydrogen bonding to the amino acid Lys58 through a structural water molecule,
Figure 3.
The three docking trials carried out on iNOS enzyme revealed that the top two scoring compounds were compounds
22 and
23 with average docking scores of −11.49 and −9.90 kcal/mol, respectively. These docking scores showed the stability of the complexes between our docked ligands and the induced nitric oxide synthase enzyme compared to MPW and 1400 W with average docking scores of −6.65 and −5.68 kcal/mol, respectively. The highest scoring compound
22 is able to fit inside the iNOS binding cavity and perform several hydrogen bonds to the key residue Glu371. Additionally, the compound interacts with the hydrophobic amino acids Met114, Trp188, and Trp457,
Figure 4.
3.3. Drug Likeness Analysis
In order to investigate the potential of the compounds identified in E. divinorum extract to serve as drug leads, we ran an extensive physicochemical property and drug likeness analysis based on the most used measures, namely Lipinski, Veber, Egan, Ghose, and Muegge.
Lipinski set five criteria for a compound to act as a drug lead: a molecular weight of no more than 500, no more than 5 H-bond donors, no more than 10 H-bond acceptors, and log-P of no more than 5. Compounds showing not more than one violation of these criteria can be promising drug candidates. Veber considered two other criteria for a compound to show good oral absorption: topological polar surface area (TPSA) and the number of rotatable bonds. TPSA is the sum of the surfaces of all the polar atoms in a given compound. According to Veber, a compound with TPSA ≤ 140 A
2 and less than 10 rotatable bonds can be absorbed well after oral administration and able to reach its molecular target. Egan stated that compounds with TPSA less than 132 A
2 and log-P value between −1 and 6 can show good orally bioavailability and serve as drug leads. The bioavailability score and sp
3 carbon fraction are two other measures of oral bioavailability. The former indicates the ability of a compound to be more than 10% bioavailable in absorption assays, while the latter is an indication of the compound’s flexibility degree. Highly flexible compounds do not usually have good oral bioavailability as they are less planar and have a complex 3D shape. Generally, compounds complying with the Lipinski rule with a bioavailability score of 0.55–0.85 and Csp
3 score between 0.25 and 1 are considered orally bioavailable [
31].
Muegge utilized simple structural rules to set a pharmacophore point filter to distinguish drug-like from non-drug-like compounds. According to this filter, a possible drug-like candidate should have log-P value between −2 and 5, a maximum TPSA of 150 A
2, less than 5 H-bond donors, less than 10 H-bond acceptors, less than 15 rotatable bonds, less than 7 rings, at least 4 carbon atoms and 1 heteroatom, and a molecular weight ranging from 200 to 400. Finally, Ghose based his drug likeness criteria on four parameters: a molecular weight between 160 and 480, a log-P value between −0.4 and 5.6, a molar refractivity between 40 and 13o, and a total atom number between 20 and 70 [
31].
Table 6 shows that among the investigated compounds, 10 showed only one or no violation of all the drug likeness criteria utilized in the physicochemical properties’ analysis. These compounds are 14, 17, 21, 24, 27, 29, 30, 35. All of the eight compounds were shown to be of high GI absorption (except 14, 17, and 30), good synthetic accessibility, and not able to inhibit the different cytochrome enzyme systems; thus, there was a low chance for possible drug–drug interactions, and they were not able to penetrate the blood–brain barrier; thus a low possibility for central adverse reactions,
Table 7. In view of this thorough analysis, these compounds can be presented as good drug leads of natural origin.
Moreover, drug likeness analysis showed that six compounds only are expected to inhibit Cyp3A4 only among the different cytochromes investigated. The rest of the compounds did not show potential to interfere with any of the cytochrome enzymes. This indicates that the extract’s components might show minimal potential drug–drug and drug–food interactions.




 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}