

Target Metabolites to Slow Down Progression of Amyotrophic Lateral Sclerosis in Mice

Abstract
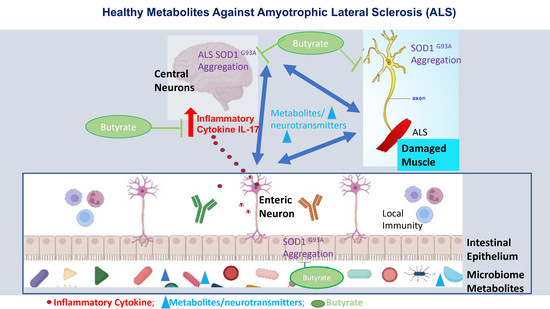
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Butyrate Treatment in Mice
2.3. Fecal Sample Collection
2.4. Bioinformatic Analysis of Metabolomics Data via Metabolon Platform
2.4.1. Sample Accessioning and Sample Preparation
2.4.2. QA/QC
2.4.3. Data Extraction and Compound Identification
2.4.4. Curation
2.4.5. Metabolite Quantification and Data Normalization
2.4.6. Immunofluorescence
2.4.7. Mouse Cytokines
2.4.8. LPS Detection
2.4.9. Statistical Analysis
3. Results
3.1. Altered Microbial Metabolites in the ALS Mice over Course of Disease
3.1.1. Changes in Carbohydrates in the SOD1G93A Mice
3.1.2. Changes of Amino Acids/Proteins in SOD1G93A Mice
3.1.3. Changes in Lipids in SOD1G93A Mice
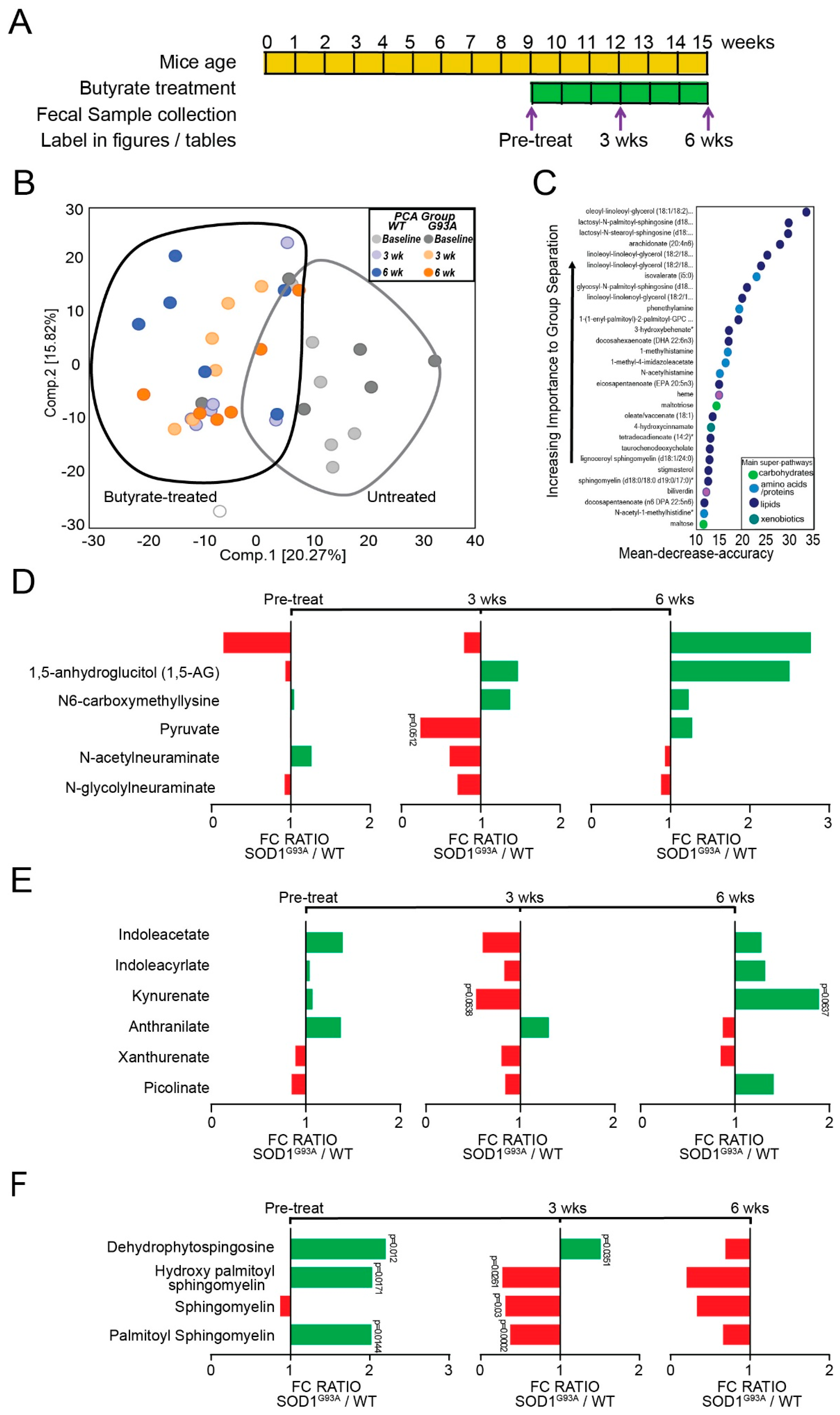
3.2. Dietary Butyrate Treatment Altered the Metabolite Profile of ALS Mice over Disease Course
3.2.1. Changes in Carbohydrates in Butyrate-Treated SOD1G93A Mice
3.2.2. Changes in Amino Acids/Proteins in Butyrate-Treated SOD1G93A Mice
3.2.3. Changes in Lipids in Butyrate Treated SOD1G93A Mice
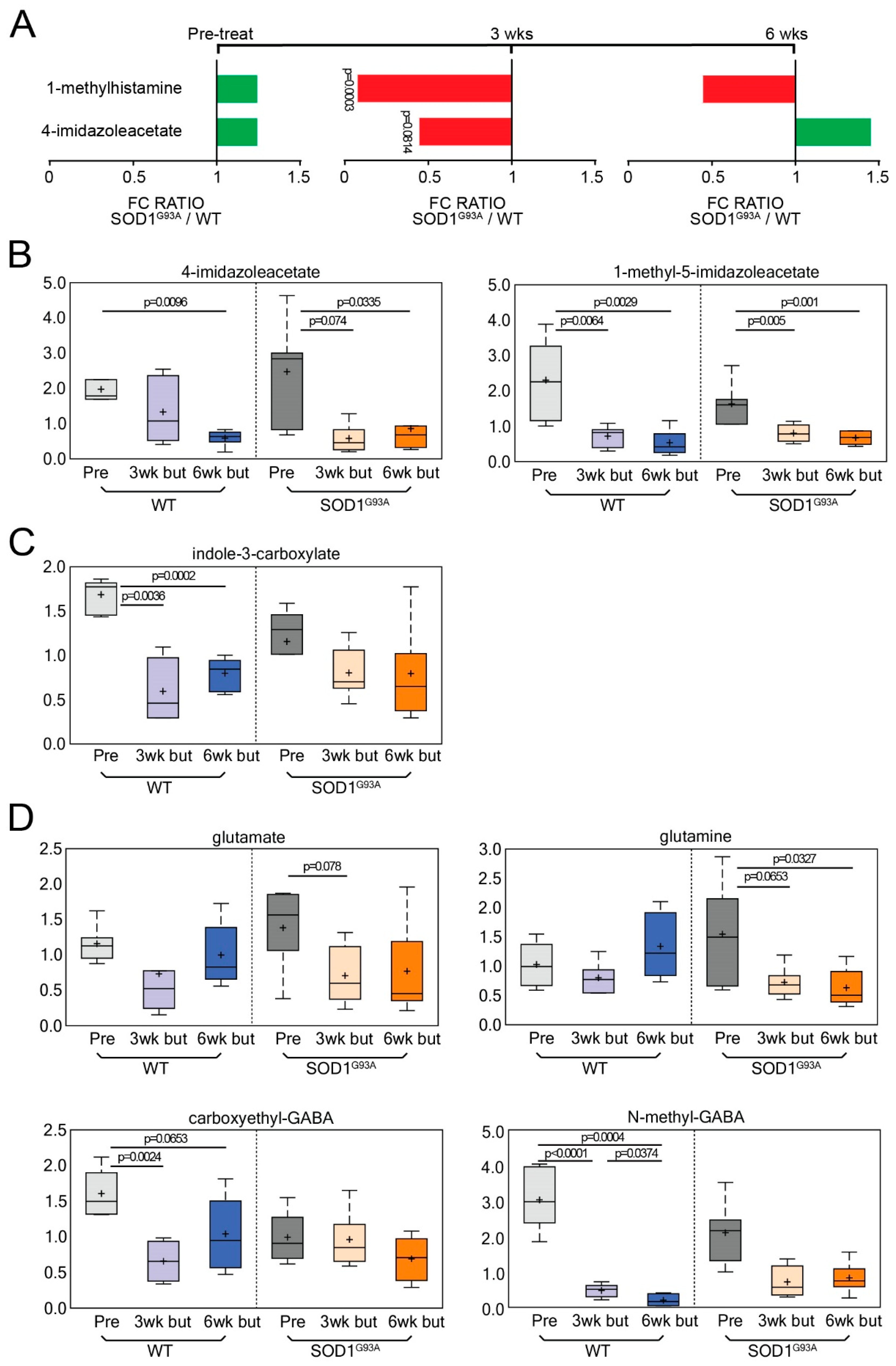
3.3. Neuroactive Metabolites Increase Significantly at Onset in ALS Mice
3.4. Dietary Butyrate Treatment Altered Neuroactive Metabolites in ALS Mice over Disease Course
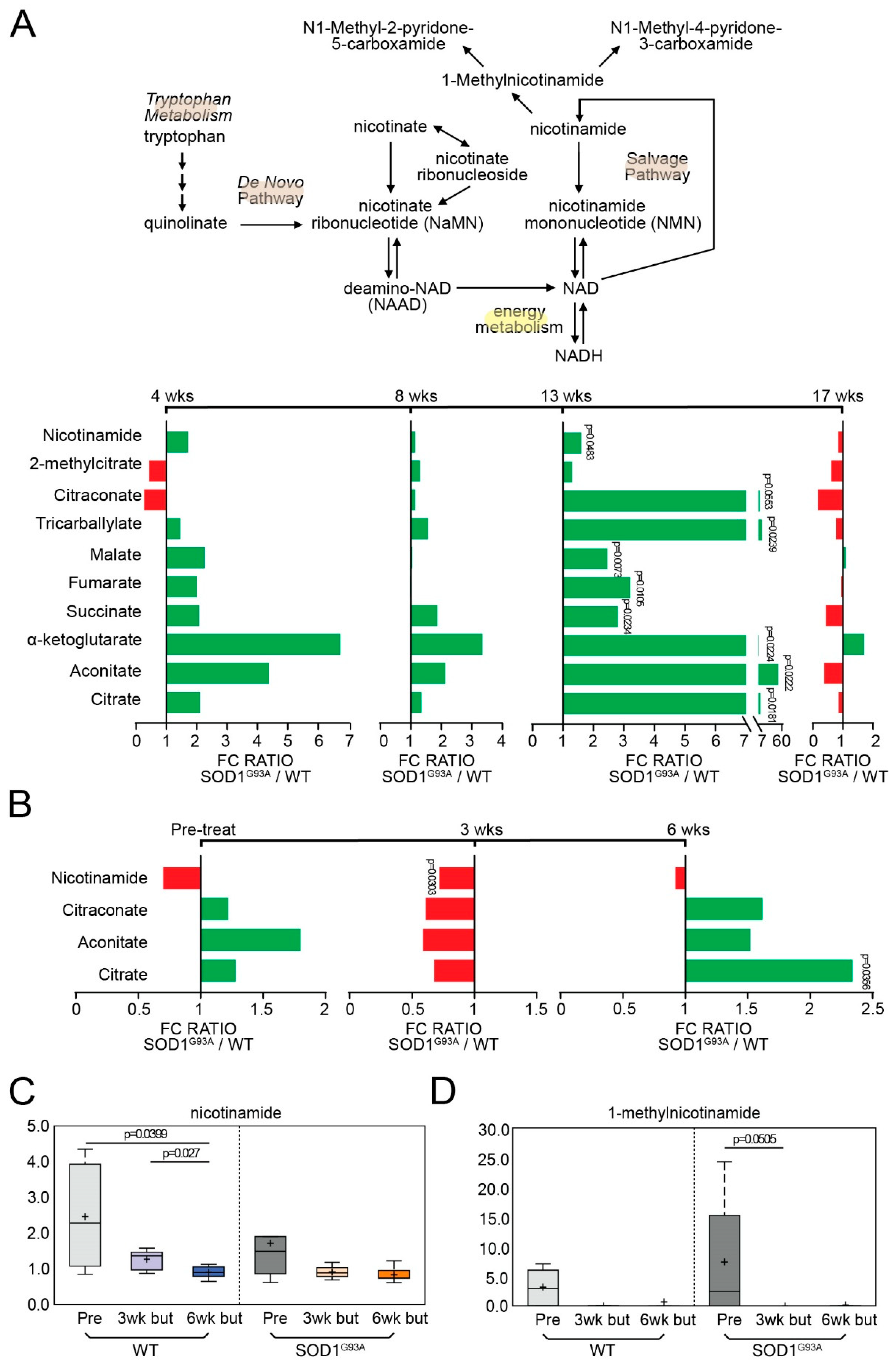
3.5. Energy-Related Metabolites Are Altered during Onset and Decreased following Butyrate Treatment in ALS Mice
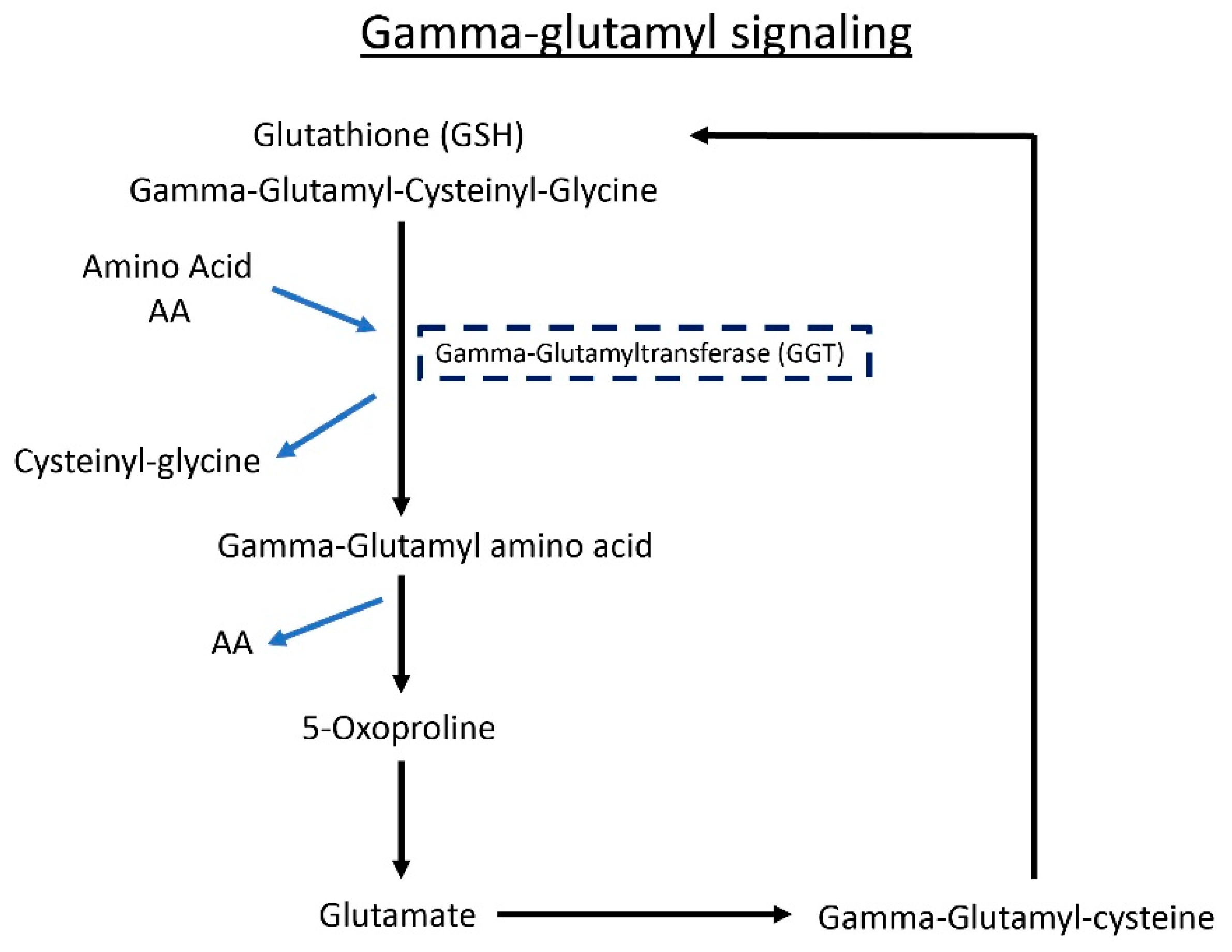
3.6. Gamma-Glutamyl Amino Acid Metabolites Are Altered following Butyrate Treatment in ALS Mice
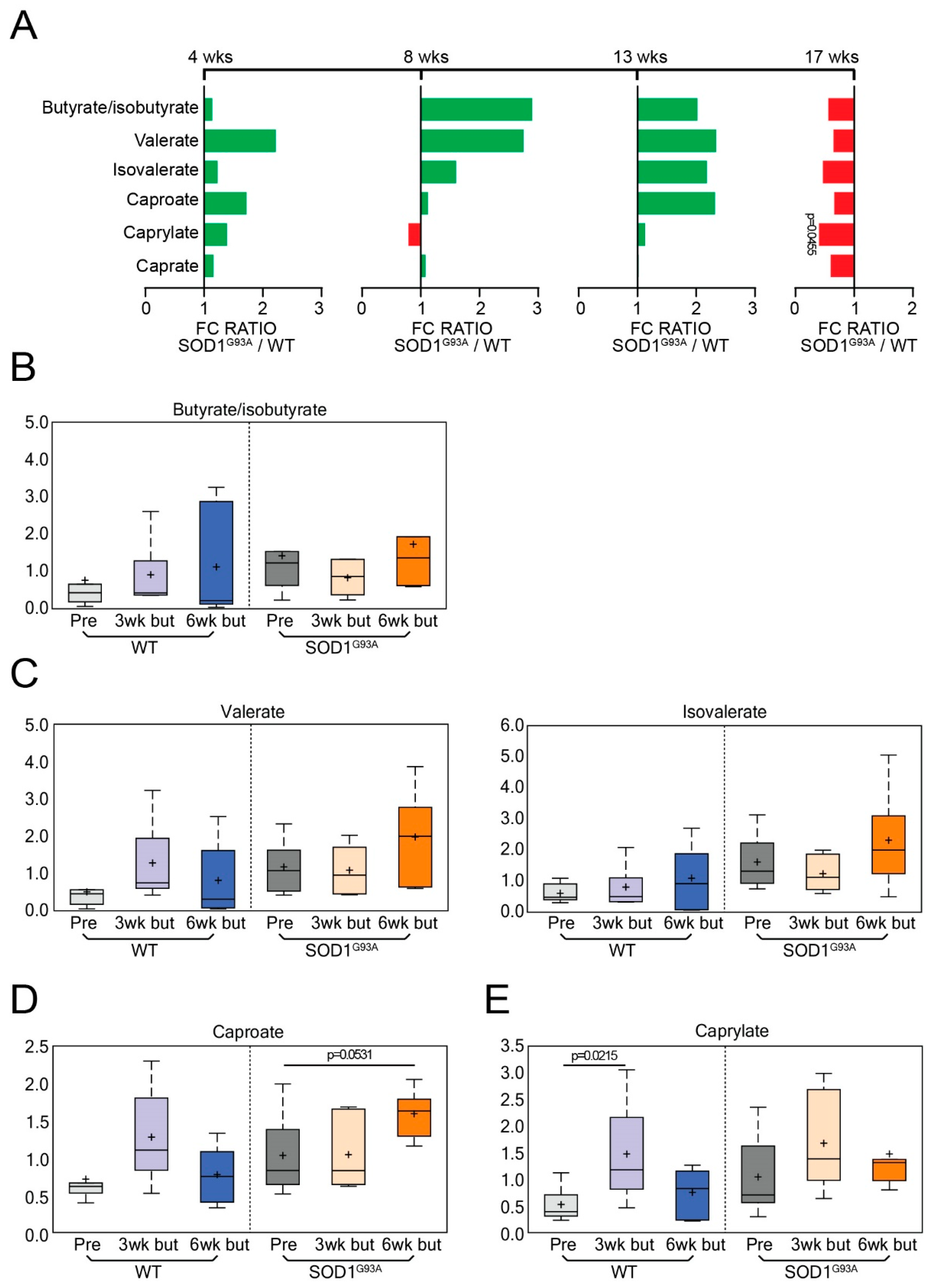
3.7. Short-Chain Fatty Acids (SCFA) in the WT and ALS Mice with or without Butyrate Treatment
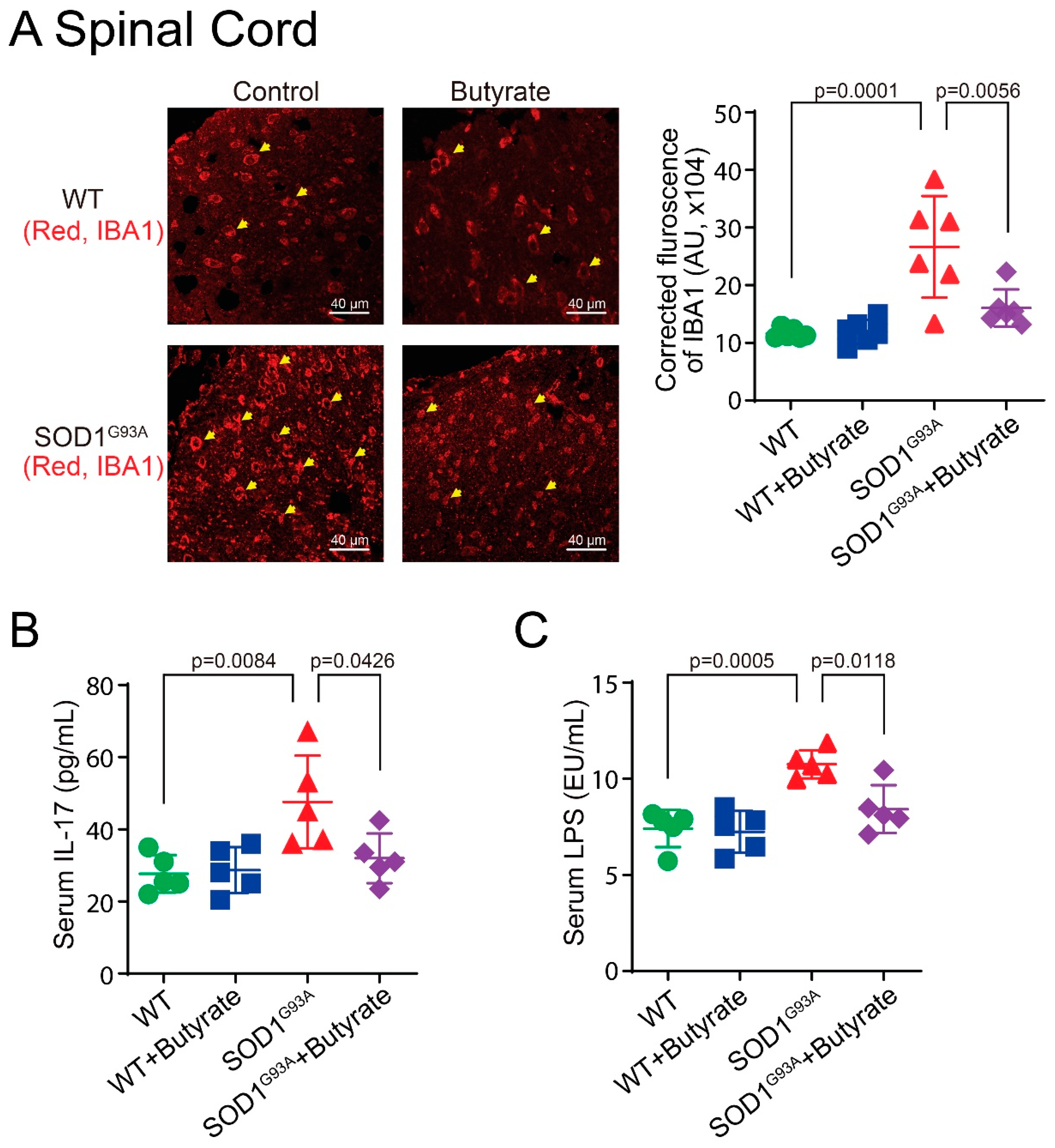
3.8. Butyrate Treatment Reduced Lumbar Spine IBA1 Expression, Serum IL-17, and LPS Expression in SOD1G93A Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensimon, G.; Lacomblez, L.; Meininger, V.; ALS/Riluzole Study Group. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Writing, G.; Edaravone, A.L.S.S.G. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.G.; Ogbu, D.; Garrett, S.; Xia, Y.; Sun, J. Aberrant enteric neuromuscular system and dysbiosis in amyotrophic lateral sclerosis. bioRxiv 2021, 13, 452097. [Google Scholar] [CrossRef]
- Turner, M.R.; Chohan, G.; Quaghebeur, G.; Greenhall, R.C.; Hadjivassiliou, M.; Talbot, K. A case of celiac disease mimicking amyotrophic lateral sclerosis. Nat. Clin. Pract. Neurol. 2007, 3, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.J.; Jewells, V.; Herfarth, H.; Castillo, M. White matter lesions suggestive of amyotrophic lateral sclerosis attributed to celiac disease. AJNR Am. J. Neuroradiol. 2010, 31, 880–881. [Google Scholar] [CrossRef] [Green Version]
- Ham, H.; Lee, B.I.; Oh, H.J.; Park, S.H.; Kim, J.S.; Park, J.M.; Cho, Y.S.; Choi, M.G. A case of celiac disease with neurologic manifestations misdiagnosed as amyotrophic lateral sclerosis. Intest. Res. 2017, 15, 540–542. [Google Scholar] [CrossRef] [Green Version]
- Gadoth, A.; Nefussy, B.; Bleiberg, M.; Klein, T.; Artman, I.; Drory, V.E. Transglutaminase 6 antibodies in the serum of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2015, 72, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef]
- Tortelli, R.; Zecca, C.; Piccininni, M.; Benmahamed, S.; Dell’Abate, M.T.; Barulli, M.R.; Capozzo, R.; Battista, P.; Logroscino, G. Plasma inflammatory cytokines are elevated in ALS. Front. Neurol. 2020, 11, 552295. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, H.; Corcia, P.; Pradat, P.F.; Bocca, C.; Gordon, P.H.; Veyrat-Durebex, C.; Mavel, S.; Nadal-Desbarats, L.; Moreau, C.; Devos, D.; et al. Metabolomics in cerebrospinal fluid of patients with amyotrophic lateral sclerosis: An untargeted approach via high-resolution mass spectrometry. J. Proteome Res. 2013, 12, 3746–3754. [Google Scholar] [CrossRef]
- Donatti, A.; Canto, A.M.; Godoi, A.B.; da Rosa, D.C.; Lopes-Cendes, I. Circulating metabolites as potential biomarkers for neurological disorders-metabolites in neurological disorders. Metabolites 2020, 10, 389. [Google Scholar] [CrossRef]
- Blasco, H.; Lanznaster, D.; Veyrat-Durebex, C.; Hergesheimer, R.; Vourch, P.; Maillot, F.; Andres, C.R.; Pradat, P.F.; Corcia, P. Understanding and managing metabolic dysfunction in Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2020, 20, 907–919. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Fidler, J.A.; Tamsett, T.J.; Bao, C.; Searles, M.; Taksir, T.V.; Misra, K.; Sidman, R.L.; Cheng, S.H.; et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 10812–10817. [Google Scholar] [CrossRef] [Green Version]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol. Rep. 2017, 5, e13443. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef]
- Ferri, A.; Coccurello, R. What is “Hyper” in the ALS hypermetabolism? Mediat. Inflamm. 2017, 2017, 7821672. [Google Scholar] [CrossRef]
- Blasco, M.P.; Chauhan, A.; Honarpisheh, P.; Ahnstedt, H.; d’Aigle, J.; Ganesan, A.; Ayyaswamy, S.; Blixt, F.; Venable, S.; Major, A.; et al. Age-dependent involvement of gut mast cells and histamine in post-stroke inflammation. J. Neuroinflammation 2020, 17, 160. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wu, S.; Liu, X.; Xia, Y.; Zhang, Y.G.; Sun, J. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PLoS ONE 2010, 5, e10505. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Binienda, A.; Twardowska, A.; Makaro, A.; Salaga, M. Dietary carbohydrates and lipids in the pathogenesis of leaky gut syndrome: An overview. Int. J. Mol. Sci. 2020, 21, 8368. [Google Scholar] [CrossRef]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fifita, J.A.; Chan Moi Fat, S.; McCann, E.P.; Williams, K.L.; Twine, N.A.; Bauer, D.C.; Rowe, D.B.; Pamphlett, R.; Kiernan, M.C.; Tan, V.X.; et al. Genetic analysis of tryptophan metabolism genes in sporadic amyotrophic lateral sclerosis. Front. Immunol. 2021, 12, 701550. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Snelson, M.; Coughlan, M.T. Dietary advanced glycation end products: Digestion, metabolism and modulation of gut microbial ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Ghazalpour, A.; Cespedes, I.; Bennett, B.J.; Allayee, H. Expanding role of gut microbiota in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 141–147. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Verdile, V.; Morello, G.; Iemmolo, R.; Aronica, E.; Cavallaro, S.; Volonté, C. Histamine regulates the inflammatory profile of SOD1-G93A microglia and the histaminergic system is dysregulated in amyotrophic lateral sclerosis. Front. Immunol. 2017, 8, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan metabolism and gut-brain homeostasis. Int. J. Mol. Sci. 2021, 22, 2973. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N. Biogenic amines: Signals between commensal microbiota and gut physiology. Front. Endocrinol. 2019, 10, 504. [Google Scholar] [CrossRef] [Green Version]
- De Moura, M.B.; Dos Santos, L.S.; Van Houten, B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environ. Mol. Mutagen. 2010, 51, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.T.; Cresci, G.A.M. The immunomodulatory functions of butyrate. J. Inflamm. Res. 2021, 14, 6025–6041. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Chaix, A.; Yooseph, S.; Panda, S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014, 20, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.; Herzog, C.; Pacheco, J.A.; Fujisaka, S.; Bullock, K.; Clish, C.B.; Kahn, C.R. Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol. Psychiatry 2018, 23, 2287–2301. [Google Scholar] [CrossRef]
- Hellwig, M.; Bunzel, D.; Huch, M.; Franz, C.M.; Kulling, S.E.; Henle, T. Stability of individual maillard reaction products in the presence of the human colonic microbiota. J. Agric. Food Chem. 2015, 63, 6723–6730. [Google Scholar] [CrossRef]
- Vaisman, N.; Lusaus, M.; Nefussy, B.; Niv, E.; Comaneshter, D.; Hallack, R.; Drory, V.E. Do patients with amyotrophic lateral sclerosis (ALS) have increased energy needs? J. Neurol. Sci. 2009, 279, 26–29. [Google Scholar] [CrossRef]
- Funalot, B.; Desport, J.C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bouteloup, C.; Desport, J.C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Oudart, H.; René, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, H.G.; Debska-Vielhaber, G.; Sander, K.; Angenstein, F.; Ludolph, A.C.; Hilfert, L.; Willker, W.; Leibfritz, D.; Heinze, H.J.; Kunz, W.S.; et al. Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2007, 25, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic analysis of serum by (1) H NMR spectroscopy in amyotrophic lateral sclerosis. Clin. Chim. Acta 2010, 411, 563–567. [Google Scholar] [CrossRef]
- Blasco, H.; Corcia, P.; Moreau, C.; Veau, S.; Fournier, C.; Vourc’h, P.; Emond, P.; Gordon, P.; Pradat, P.F.; Praline, J.; et al. 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e13223. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [Green Version]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted metabolomics yields insight into ALS disease mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef]
- Valbuena, G.N.; Cantoni, L.; Tortarolo, M.; Bendotti, C.; Keun, H.C. Spinal cord metabolic signatures in models of fast- and slow-progressing SOD1(G93A) amyotrophic lateral sclerosis. Front. Neurosci. 2019, 13, 1276. [Google Scholar] [CrossRef]
- Bame, M.; Grier, R.E.; Needleman, R.; Brusilow, W.S. Amino acids as biomarkers in the SOD1(G93A) mouse model of ALS. Biochim. Biophys. Acta 2014, 1842, 79–87. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115, 1249–1273. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Diether, N.E.; Willing, B.P. Microbial fermentation of dietary protein: An important factor in diet-microbe-host interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.; Fonseca, S.; Carding, S.R. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 2020, 11, 135–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, V.X.; Guillemin, G.J. Kynurenine pathway metabolites as biomarkers for amyotrophic lateral sclerosis. Front. Neurosci. 2019, 13, 1013. [Google Scholar] [CrossRef] [PubMed]
- Foster, T. Staphylococcus. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Yu, B.; Zheng, Y.; Alexander, D.; Morrison, A.C.; Coresh, J.; Boerwinkle, E. Genetic determinants influencing human serum metabolome among African Americans. PLoS Genet. 2014, 10, e1004212. [Google Scholar] [CrossRef]
- Di Gioia, D.; Bozzi Cionci, N.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggìa, F.; Lucenti, M.A.; Bersano, E.; Cantello, R.; et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef]
- Zeng, Q.; Shen, J.; Chen, K.; Zhou, J.; Liao, Q.; Lu, K.; Yuan, J.; Bi, F. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 2020, 10, 12998. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Zhang, R.; Hadlock, K.G.; Do, H.; Yu, S.; Honrada, R.; Champion, S.; Forshew, D.; Madison, C.; Katz, J.; Miller, R.G.; et al. Gene expression profiling in peripheral blood mononuclear cells from patients with sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2011, 230, 114–123. [Google Scholar] [CrossRef]
- Fang, X.; Wang, X.; Yang, S.; Meng, F.; Wang, X.; Wei, H.; Chen, T. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front. Microbiol. 2016, 7, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, K.; Hachiya, N.S. Hypothesis: Gut as source of motor neuron toxin in the development of ALS. Med. Hypotheses 2006, 66, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Longstreth, W.T., Jr.; Meschke, J.S.; Davidson, S.K.; Smoot, L.M.; Smoot, J.C.; Koepsell, T.D. Hypothesis: A motor neuron toxin produced by a clostridial species residing in gut causes ALS. Med. Hypotheses 2005, 64, 1153–1156. [Google Scholar] [CrossRef]
- Toepfer, M.; Folwaczny, C.; Klauser, A.; Riepl, R.L.; Muller-Felber, W.; Pongratz, D. Gastrointestinal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 1999, 1, 15–19. [Google Scholar]
- Martin, S.; Battistini, C.; Sun, J. A gut feeling in amyotrophic lateral sclerosis: Microbiome of mice and men. Front. Cell Infect. Microbiol. 2022, 12, 839526. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Comparisons | ||||
|---|---|---|---|---|
| Welch’s Two-Sample t-Test | SOD1G93A/WT | |||
| 4 Week | 8 Week | 13 Week | 17 Week | |
| Total biochemicals p ≤ 0.05 | 61 | 7 | 180 | 10 |
| Biochemicals (↑↓) | 49 | 12 | 5 | 2 | 175 | 5 | 6 | 4 |
| Statistical Comparisons | |||
|---|---|---|---|
| Welch’s Two-Sample t-Test | SOD1G93A/WT | ||
| Baseline | 3 Week Post Butyrate Treatment | 6 Week Post Butyrate Treatment | |
| Total biochemicals p ≤ 0.05 | 74 | 107 | 81 |
| Biochemicals (↑↓) | 61 | 13 | 43 | 64 | 32 | 49 |
| WT | G93A | |||
|---|---|---|---|---|
| Biochemical Name | 3 Week Post-Butyrate Treatment /Baseline | 6 Week Post-Butyrate Treatment /Baseline | 3 Week Post-Butyrate Treatment /Baseline | 6 Week Post-Butyrate Treatment /Baseline |
| Gamma-glutamylalanine | 1.34 | 0.59 | 0.51 | 0.92 |
| Gamma-glutamylglutamate | 0.49 | 0.33 | 0.26 | 0.43 |
| Gamma-glutamylglutamine | 0.79 | 0.78 | 0.30 | 0.42 |
| Gamma-glutamylglycine | 0.86 | 0.49 | 0.39 | 0.69 |
| Gamma-glutamylhistidine | 0.48 | 0.50 | 0.33 | 0.41 |
| Gamma-glutamylisoleucine | 0.81 | 0.61 | 0.37 | 0.61 |
| Gamma-glutamylleucine | 1.30 | 0.91 | 0.36 | 0.64 |
| Gamma-glutamyl-apha-lysine | 0.90 | 0.60 | 0.37 | 0.58 |
| Gamma-glutamyl-epsilon-lysine | 1.39 | 1.84 | 0.37 | 0.46 |
| Gamma-glutamylmethionine | 1.43 | 1.16 | 0.25 | 0.60 |
| Gamma-glutamylphenyalanine | 0.88 | 0.62 | 0.30 | 0.57 |
| Gamma-glutamylthreonine | 0.98 | 0.71 | 0.35 | 0.69 |
| Gamma-glutamyltryosine | 0.73 | 0.72 | 0.33 | 0.75 |
| Gamma-glutamylvaline | 1.20 | 0.74 | 0.32 | 0.59 |
| Gamma-glutamylserine | 0.57 | 0.56 | 0.44 | 0.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogbu, D.; Zhang, Y.; Claud, K.; Xia, Y.; Sun, J. Target Metabolites to Slow Down Progression of Amyotrophic Lateral Sclerosis in Mice. Metabolites 2022, 12, 1253. https://doi.org/10.3390/metabo12121253
Ogbu D, Zhang Y, Claud K, Xia Y, Sun J. Target Metabolites to Slow Down Progression of Amyotrophic Lateral Sclerosis in Mice. Metabolites. 2022; 12(12):1253. https://doi.org/10.3390/metabo12121253
Chicago/Turabian StyleOgbu, Destiny, Yongguo Zhang, Katerina Claud, Yinglin Xia, and Jun Sun. 2022. "Target Metabolites to Slow Down Progression of Amyotrophic Lateral Sclerosis in Mice" Metabolites 12, no. 12: 1253. https://doi.org/10.3390/metabo12121253