
Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus epidermidis RP62A

 , ,
, ,  and
and
Abstract
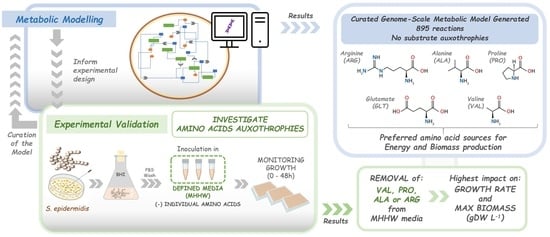
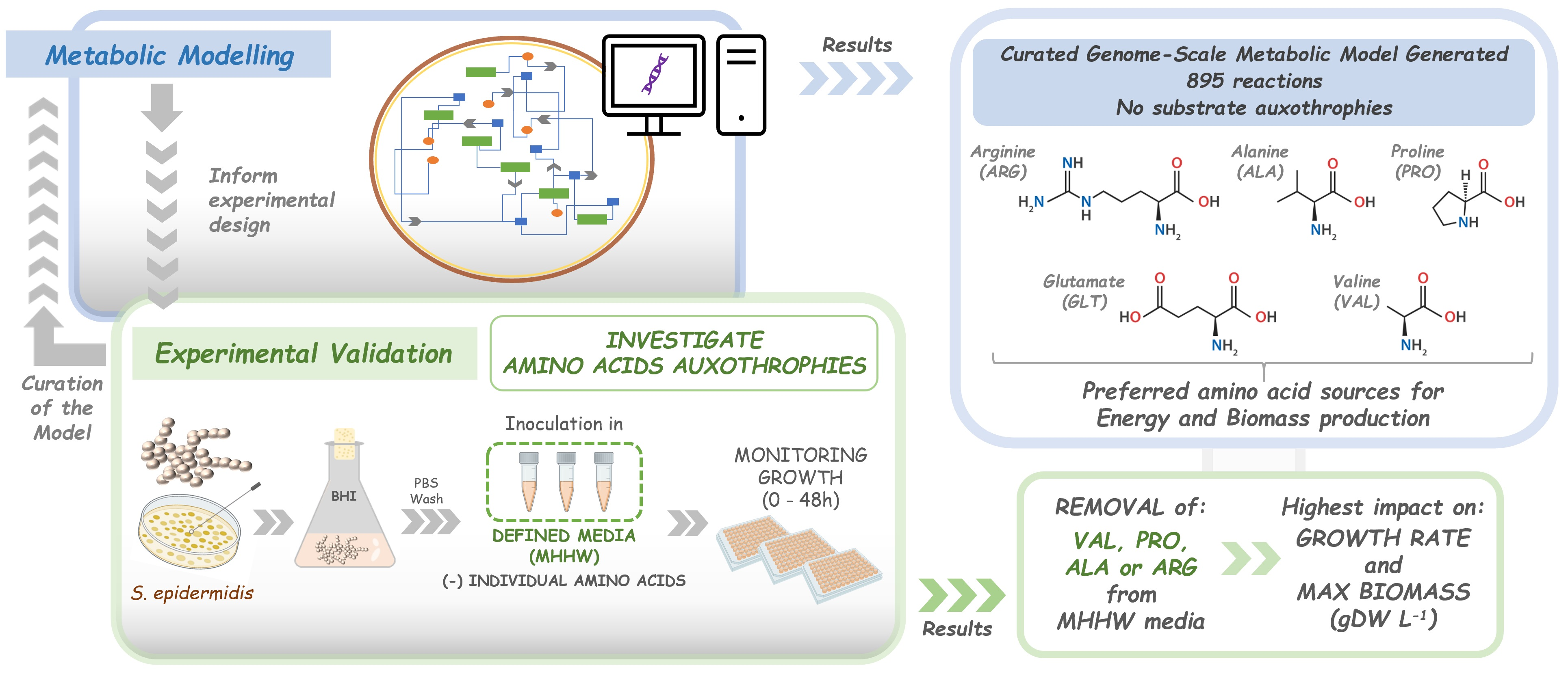
:
1. Introduction
2. Materials and Methods
2.1. Model Construction
- The top level
- module, which serves to import the modules listed below.
- Automatically generated
- reactions extracted from the PGDB, automatically corrected where necessary, as described in Section 2.2.
- Transport reactions
- to account for the import of the various media components and export of metabolic by-products (Section 2.4.1).
- Biomass generation
- consisting of “pseudo-transporters” to allow for the export of biomass precursors. Biomass composition, comprised of biofilm and planktonic cell composition, was defined as a modification of that described for S. aureus [23] (Supplementary File SI).
- Electron Transport Chain/Oxidative Phosphorylation
- Additional reactions
- found to be necessary for the synthesis of biomass precursors, not present in the PGDB. Candidate reactions were included after confirming the presence of the genes encoding the corresponding enzymes or other experimental evidence in updated versions of BioCyc, the KEGG database for RP62A, and biochemical databases for Staphylococcus spp.
2.2. Model Curation and Theoretical Validation
2.3. Model Analysis
Linear Programming Assumptions and Constraints
2.4. Experimental Conditions
2.4.1. Defined Rich Media Design
2.4.2. Inoculum and Bacterial Strains
2.4.3. Impact on Growth in the Removal of Individual Amino Acids
2.4.4. Growth Parameter Calculation
3. Results
3.1. Model: General Properties
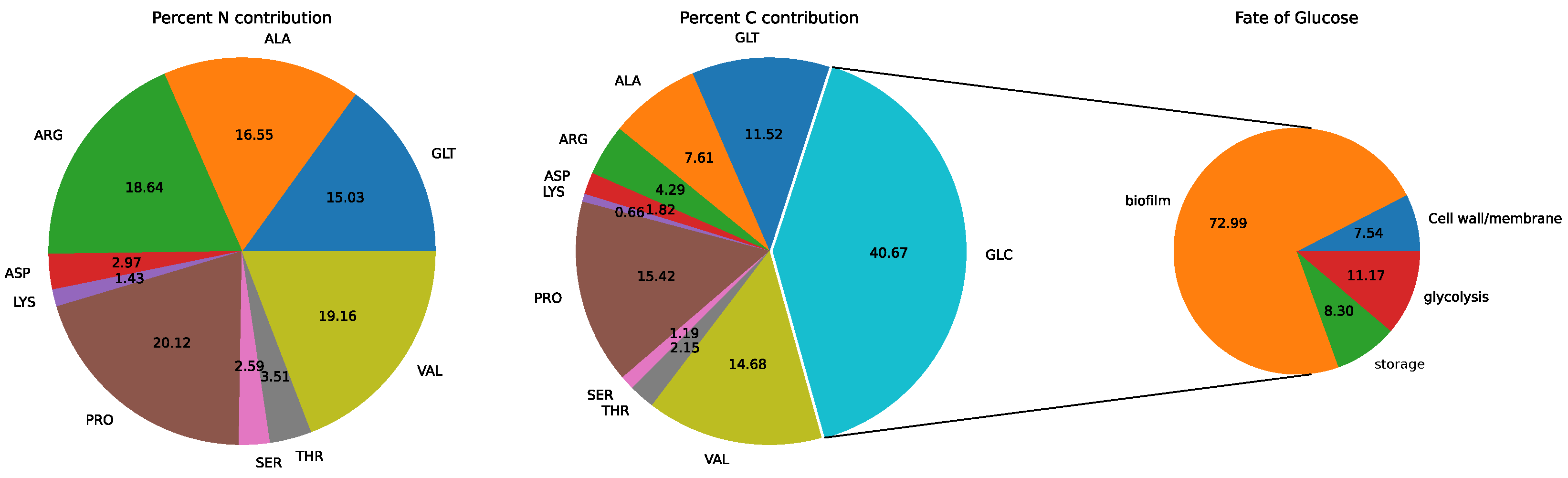
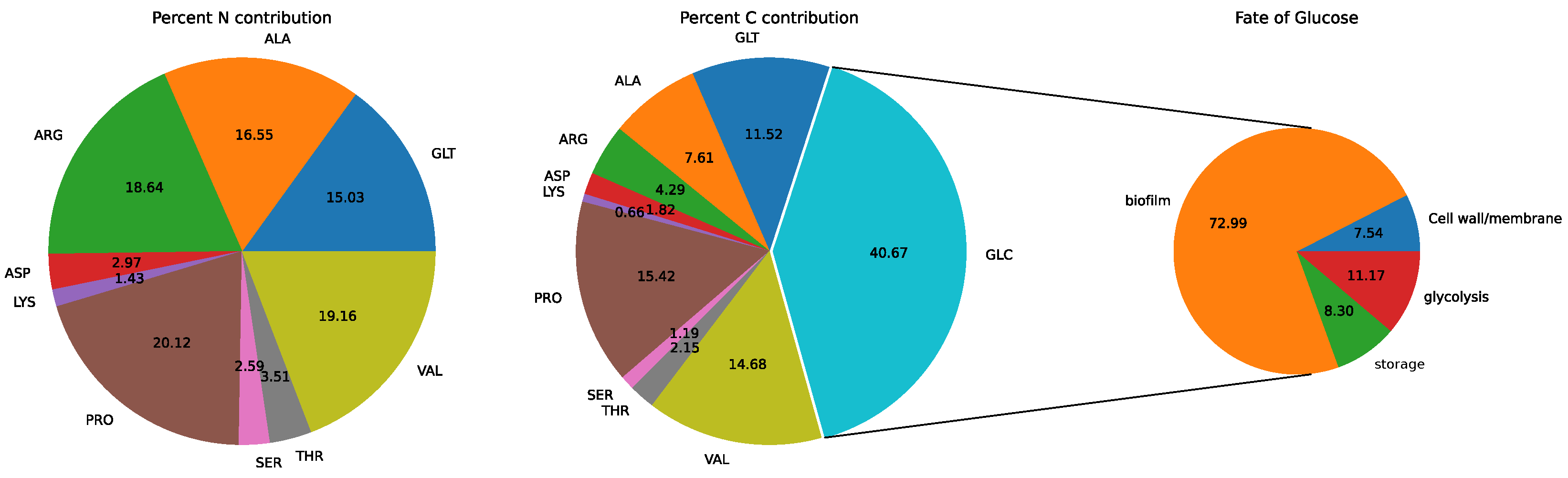
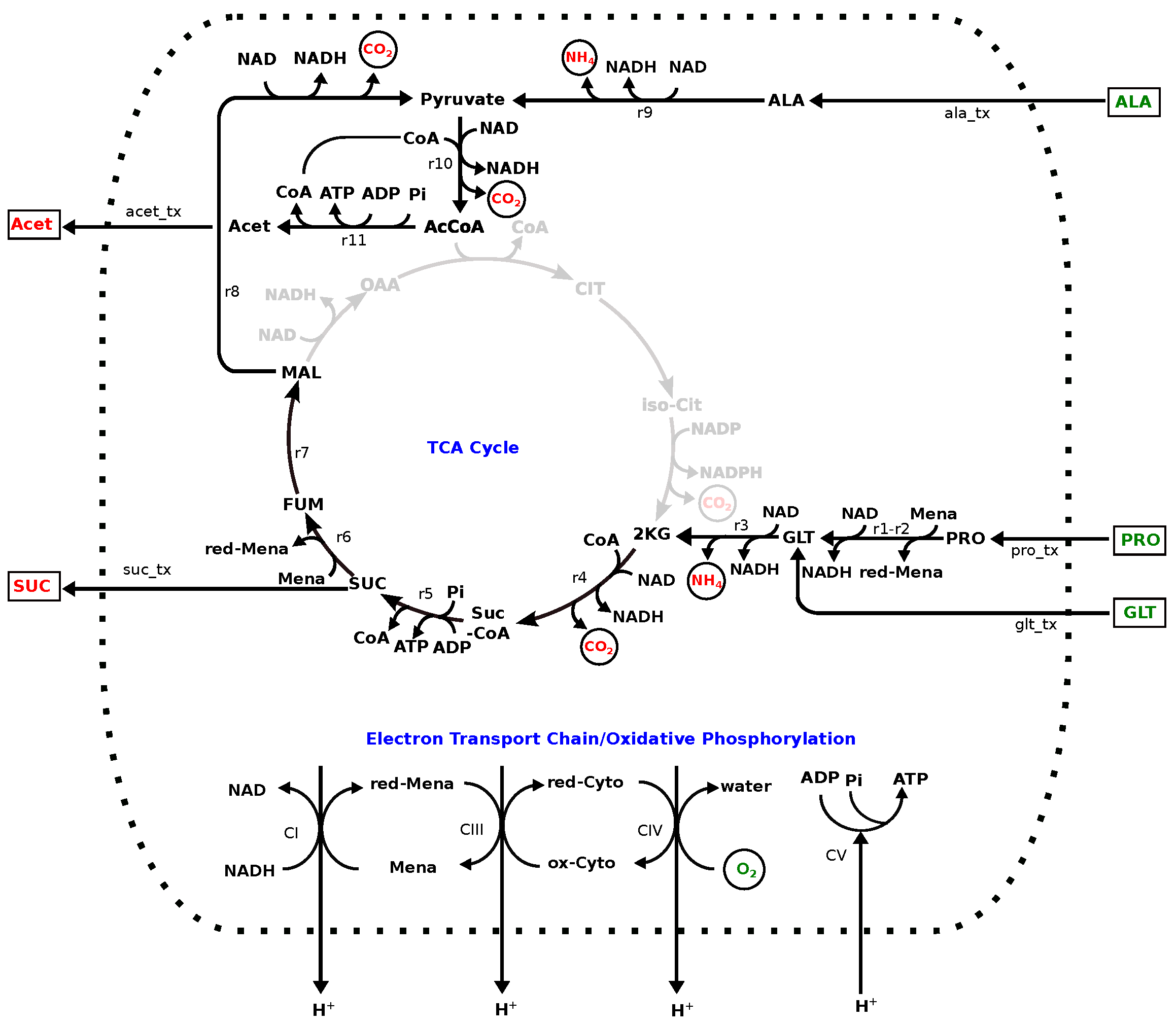
3.2. Model: Growth on MHHW Medium
3.3. Impact of Removing Individual Amino Acids
3.3.1. Model
3.3.2. Experimental
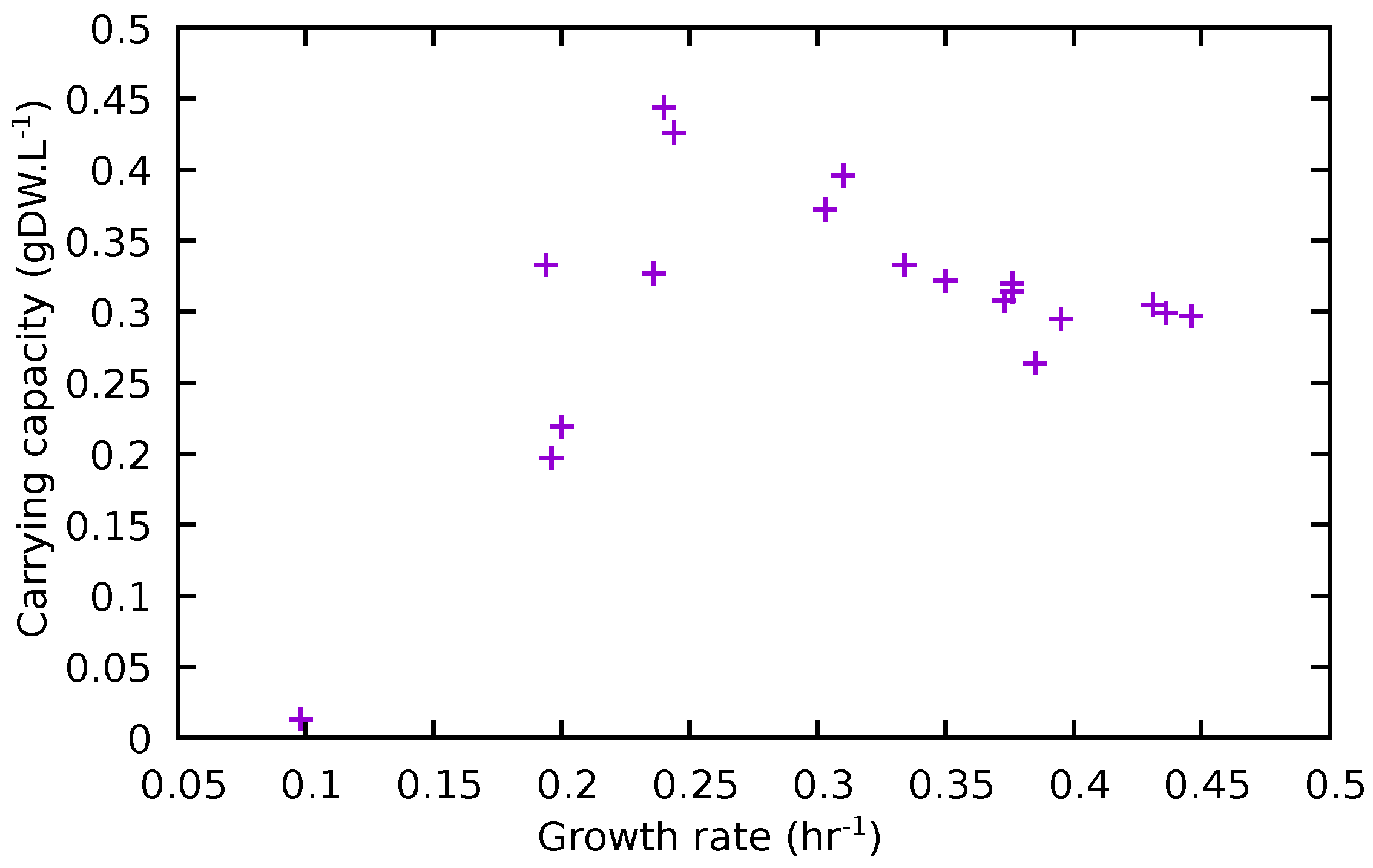
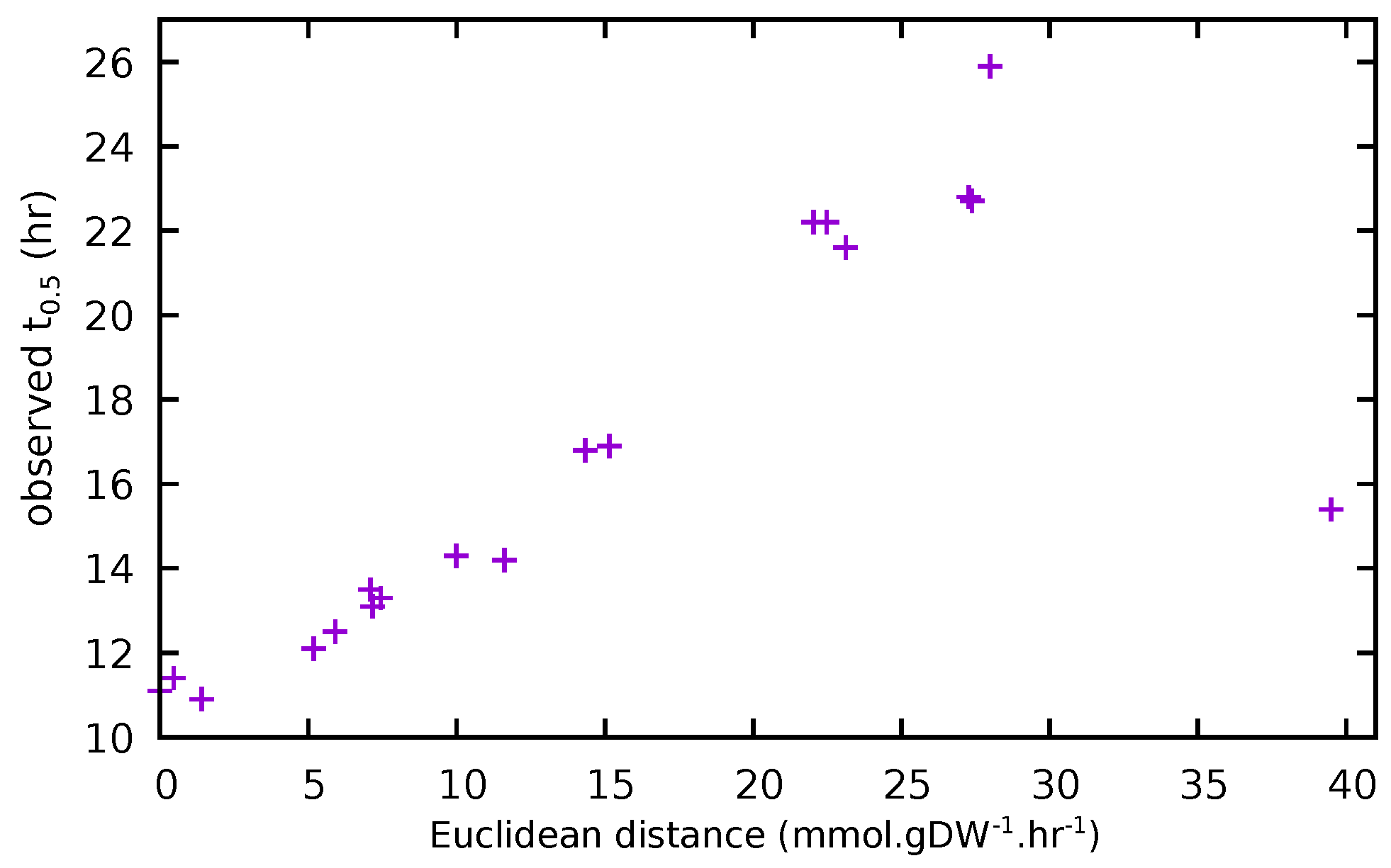
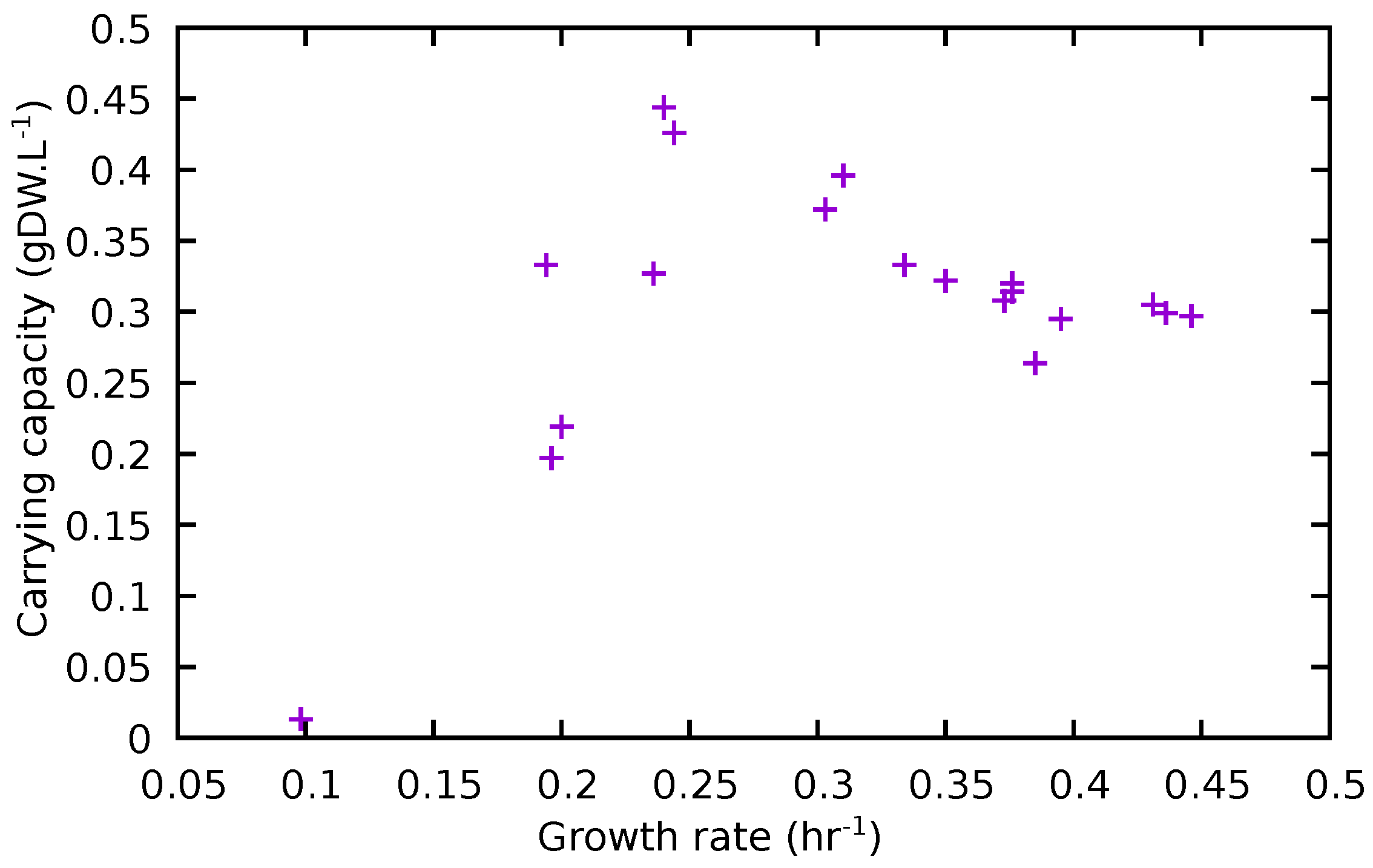
3.4. Comparison of Experimental and Model Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pereira, A.T. Coagulase-negative strains of staphylococcus possessing antigen 51 as agents of urinary infection. J. Clin. Pathol. 1962, 15, 252–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [Green Version]
- Argemi, X.; Riegel, P.; Lavigne, T.; Lefebvre, N.; Grandpré, N.; Hansmann, Y.; Jaulhac, B.; Prévost, G.; Schramm, F. Implementation of matrix-assisted laser desorption ionization–time of flight mass spectrometry in routine clinical laboratories improves identification of coagulase-negative staphylococci and reveals the pathogenic role of Staphylococcus lugdunensis. J. Clin. Microbiol. 2015, 53, 2030–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, M. Staphylococcus epidermidis—The ‘accidental’ pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Ni, J.; Shang, F.; Chen, X.; Zhang, M. Autoinducer-2 increases biofilm formation via an ica- and bhp-dependent manner in Staphylococcus epidermidis RP62A. Microbes Infect. 2015, 17, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Somerville, G.A.; Proctor, R.A. At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol. Mol. Biol. Rev. 2009, 73, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Strasters, K.C.; Winkler, K.C. Carbohydrate metabolism of Staphylococcus aureus. J. Gen. Microbiol. 1963, 33, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, S.; Pané-Farré, J.; Kohler, C.; Hecker, M.; Engelmann, S. Anaerobic gene expression in Staphylococcus aureus. J. Bacteriol. 2007, 189, 4275–4289. [Google Scholar] [CrossRef] [Green Version]
- Schwan, W.R.; Wetzel, K.J.; Gomez, T.S.; Stiles, M.A.; Beitlich, B.D.; Grunwald, S. Low-proline environments impair growth, proline transport and in vivo survival of Staphylococcus aureus strain-specific putP mutants. Microbiology 2004, 150, 1055–1061. [Google Scholar] [CrossRef]
- Nuxoll, A.S.; Halouska, S.M.; Sadykov, M.R.; Hanke, M.L.; Bayles, K.W.; Kielian, T.; Powers, R.; Fey, P.D. CcpA regulates arginine biosynthesis in Staphylococcus aureus through repression of proline catabolism. PLoS Pathog 2012, 8, e1003033. [Google Scholar] [CrossRef]
- Halsey, C.R.; Lei, S.; Wax, J.K.; Lehman, M.K.; Nuxoll, A.S.; Steinke, L.; Sadykov, M.; Powers, R.; Fey, P.D. Amino acid catabolism in Staphylococcus aureus and the function of carbon catabolite repression. mBio 2017, 8, e01434-16. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gutierrez, E.; Walsh, C.J.; Sayavedra, L.; Diaz-Calvo, T.; Thapa, D.; Ruas-Madiedo, P.; Mayer, M.J.; Cotter, P.D.; Narbad, A. Genotypic and phenotypic characterization of fecal Staphylococcus epidermidis isolates suggests plasticity to adapt to different human body sites. Front. Microbiol. 2020, 11, 688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, G.I.; Utrilla, J.; Nurk, S.; Brunk, E.; Monk, J.M.; Ebrahim, A.; Palsson, B.O.; Feist, A.M. Model-driven discovery of underground metabolic functions in Escherichia coli. Proc. Natl. Acad. Sci. USA 2015, 112, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.E.; Park, S.J.; Kim, W.J.; Kim, H.; Kim, B.; Lee, H.; Shin, J.; Lee, S.Y. One-step fermentative production of aromatic polyesters from glucose by metabolically engineered Escherichia coli strains. Nat. Commun. 2018, 9, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presta, L.; Bosi, E.; Mansouri, L.; Dijkshoorn, L.; Fani, R.; Fondi, M. Constraint-based modeling identifies new putative targets to fight colistin-resistant A. baumannii infections. Sci. Rep. 2017, 7, 3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Haleem, A.M.; Hefzi, H.; Mineta, K.; Gao, X.; Gojobori, T.; Palsson, B.O.; Lewis, N.E.; Jamshidi, N. Functional interrogation of Plasmodium genus metabolism identifies species- and stage-specific differences in nutrient essentiality and drug targeting. PLoS Comput. Biol. 2018, 14, e1005895. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.; Kim, W.; Kim, T.Y.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Tejera, N.; Crossman, L.; Pearson, B.; Stoakes, E.; Nasher, F.; Djeghout, B.; Poolman, M.; Wain, J.; Singh, D. Genome-scale metabolic model driven design of a defined medium for Campylobacter jejuni M1cam. Front. Microbiol. 2020, 11, 1072. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012, 40, D742–D753. [Google Scholar] [CrossRef] [Green Version]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Briefings Bioinform. 2017, 20, 1085–1093. [Google Scholar] [CrossRef]
- Poolman, M. ScrumPy: Metabolic modelling with Python. IEE Proc.-Syst. Biol. 2006, 153, 375–378. [Google Scholar] [CrossRef]
- Ahmad, A.; Hartman, H.B.; Krishnakumar, S.; Fell, D.A.; Poolman, M.G.; Srivastava, S. A genome-scale model of Geobacillus thermoglucosidasius (C56-YS93) reveals its biotechnological potential on rice straw hydrolysate. J. Biotechnol. 2017, 251, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, M.; Kummel, A.; Ruinatscha, R.; Panke, S. In silico genome-scale reconstruction and validation of the Staphylococcus aureus metabolic network. Biotechnol. Bioeng. 2005, 92, 850–864. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, A.; Purvis, P.; Portelance, V. Role of menaquinone in nitrate respiration in Staphylococcus aureus. J. Bacteriol. 1974, 117, 911–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, K.A.; Lascelles, J. Nitrate reductase system in Staphylococcus aureus wild type and mutants. J. Bacteriol. 1975, 123, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Tynecka, Z.; Szczesniak, Z.; Malm, A.; Los, R. Energy conservation in aerobically grown Staphylococcus aureus. Res. Microbiol. 1999, 150, 555–566. [Google Scholar] [CrossRef]
- McNamara, P.J.; Proctor, R.A. Staphylococcus aureus small colony variants, electron transport and persistent infections. Int. J. Antimicrob. Agents 2000, 14, 117–122. [Google Scholar] [CrossRef]
- Gevorgyan, A.; Poolman, M.G.; Fell, D.A. Detection of stoichiometric inconsistencies in biomolecular models. Bioinformatics 2008, 24, 2245–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poolman, M.G.; Kundu, S.; Shaw, R.; Fell, D.A. Responses to light intensity in a genome-scale model of rice metabolism. Plant Physiol. 2013, 162, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzhütter, H.G. The principle of flux minimization and its application to estimate stationary fluxes in metabolic networks. Eur. J. Biochem. 2004, 271, 2905–2922. [Google Scholar] [CrossRef]
- Holzhütter, H.G. The generalized flux-minimization method and its application to metabolic networks affected by enzyme deficiencies. Biosystems 2006, 83, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Carlson, R.P.; Fell, D.A.; Poolman, M.G. Modelling metabolism of the diatom Phaeodactylum tricornutum. Biochem. Soc. Trans. 2015, 43 6, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.O. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef] [PubMed]
- Hartman, H.B.; Fell, D.A.; Rossell, S.; Jensen, P.R.; Woodward, M.J.; Thorndahl, L.; Jelsbak, L.; Olsen, J.E.; Raghunathan, A.; Daefler, S.; et al. Identification of potential drug targets in Salmonella enterica sv. Typhimurium using metabolic modelling and experimental validation. Microbiology 2014, 160, 1252–1266. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Hastings, J.; White, P. A chemically defined medium for slime production by coagulase-negative Staphylococci. J. Med. Microbiol. 1991, 34, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Jain, A. Glucose & sodium chloride induced biofilm production & ica operon in clinical isolates of staphylococci. Indian J. Med. Res. 2013, 138, 262–266. [Google Scholar] [PubMed]
- Rossatto, F.C.P.; Pinto, J.B.; Costa, G.A.; Frazzon, A.P.G. In vitro biofilm formation ability of staphylococci under different growth conditions. Int. J. Appl. Microbiol. Biotechnol. Res. 2017, 5, 12–19. [Google Scholar]
- Kofoed, E.M.; Yan, D.; Katakam, A.K.; Reichelt, M.; Lin, B.; Kim, J.; Park, S.; Date, S.V.; Monk, I.R.; Xu, M.; et al. De Novo guanine biosynthesis but not the riboswitch-regulated purine salvage pathway is required for Staphylococcus aureus Infection In Vivo. J. Bacteriol. 2016, 198, 2001–2015. [Google Scholar] [CrossRef] [Green Version]
- Goncheva, M.I.; Flannagan, R.S.; Heinrichs, D.E.; Bäumler, A.J. De novo purine biosynthesis is required for intracellular growth of Staphylococcus aureus and for the hypervirulence phenotype of a purR mutant. Infect. Immun. 2020, 88, e00104-20. [Google Scholar] [CrossRef] [Green Version]
- Paley, S.M.; Karp, P.D. The Pathway Tools cellular overview diagram and Omics Viewer. Nucleic Acids Res. 2006, 34, 3771–3778. [Google Scholar] [CrossRef] [Green Version]
- Mazharul Islam, M.; Thomas, V.C.; Van Beek, M.; Ahn, J.S.; Alqarzaee, A.A.; Zhou, C.; Fey, P.D.; Bayles, K.W.; Saha, R. An integrated computational and experimental study to investigate Staphylococcus aureus metabolism. NPJ Syst. Biol. Appl. 2020, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Seif, Y.; Monk, J.M.; Mih, N.; Tsunemoto, H.; Poudel, S.; Zuniga, C.; Broddrick, J.; Zengler, K.; Palsson, B.O. A computational knowledge-base elucidates the response of Staphylococcus aureus to different media types. PLoS Comput. Biol. 2019, 15, e1006644. [Google Scholar] [CrossRef]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B.O. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Burd, H.; Liu, J.; Almaas, E.; Wiest, O.; Barabási, A.L.; Oltvai, Z.N.; Kapatral, V. Comparative genome-scale metabolic reconstruction and flux balance analysis of multiple Staphylococcus aureus genomes identify novel antimicrobial drug targets. J. Bacteriol. 2009, 191, 4015–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, S.A.; Palsson, B.O. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: An initial draft to the two-dimensional annotation. BMC Microbiol. Electron. Resour. 2005, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Díaz Calvo, T. Investigating the Metabolism of Non-Aureus Staphylococci Relevant to Prosthetic Joint Infection. Ph.D. Dissertation, University of East Anglia, Norwich, UK, 2020. [Google Scholar]
- Reeves, R.E.; Warren, L.G.; Susskind, B.; Lo, H.S. An energy-conserving pyruvate-to-acetate pathway in Entamoeba histolytica. Pyruvate synthase and a new acetate thiokinase. J. Biol. Chem. 1977, 252, 726–731. [Google Scholar] [CrossRef]
- Lincoln, R.A.; Leigh, J.A.; Jones, N.C. The amino acid requirements of Staphylococcus aureus isolated from cases of bovine mastitis. Vet. Microbiol. 1995, 45, 275–279. [Google Scholar] [CrossRef]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.-i.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Charlier, C.; Cretenet, M.; Even, S.; Le Loir, Y. Interactions between Staphylococcus aureus and lactic acid bacteria: An old story with new perspectives. Int. J. Food Microbiol. 2009, 131, 30–39. [Google Scholar] [CrossRef]
- Li, C.; Sun, F.; Cho, H.; Yelavarthi, V.; Sohn, C.; He, C.; Schneewind, O.; Bae, T. CcpA Mediates Proline Auxotrophy and Is Required for Staphylococcus aureus Pathogenesis. J. Bacteriol. 2010, 192, 3883–3892. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shen, Z.; Tang, J.; Huang, Q.; Jian, Y.; Liu, Y.; Wang, Y.; Ma, X.; Liu, Q.; He, L.; et al. Extracellular DNA released by glycine-auxotrophic Staphylococcus epidermidis small colony variant facilitates catheter-related infections. Commun. Biol. 2021, 4, 904. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain (Publication) | Reactions | Transporters | Metabolites | Genes | Conservation | |
|---|---|---|---|---|---|---|
| Mass | Energy | |||||
| RP62A (this work) | 895 | 95 | 864 | 611 | yes | yes |
| iSA863 [41] | 1545 | NR | 1379 | NR | yes * | yes |
| iYS854 [42] | 1440 | NR | 1327 | NR | NR | yes |
| multiple [43] | 1475 | NR | 1232 | NR | NR | NR |
| multiple [44] | 1497 | 146 | 1431 | NR | NR | NR |
| iMH551 [23] | 774 | 92 | 712 | 726 | NR | yes |
| iSB619 [45] | 640 | 84 | 571 | 581 | no | NR |
| Removed | Impact | Removed | Impact | Removed | Impact | Removed | Impact |
|---|---|---|---|---|---|---|---|
| None | 0 | ASN | 0.462 | ASP | 1.41 | SER | 5.19 |
| LYS | 5.91 | GLY | 7.10 | ILE | 7.17 | MET | 7.44 |
| CYS | 10.0 | HIS | 11.6 | TYR | 14.3 | PHE | 15.2 |
| THR | 22.0 | LEU | 22.5 | GLT | 23.1 | ARG | 27.3 |
| ALA | 27.4 | TRP | 28.0 | PRO | 39.5 | VAL | 41.0 |
| Removed | K | t0.5 | Removed | K | t0.5 | ||
|---|---|---|---|---|---|---|---|
| None | 0.436 | 0.299 | 11.1 | ASN | 0.431 | 0.305 | 11.4 |
| ASP | 0.446 | 0.297 | 10.9 | SER | 0.395 | 0.295 | 12.1 |
| LYS | 0.385 | 0.264 | 12.5 | GLY | 0.376 | 0.320 | 13.5 |
| ILE | 0.376 | 0.314 | 13.1 | MET | 0.373 | 0.308 | 13.3 |
| CYS | 0.350 | 0.322 | 14.3 | HIS | 0.334 | 0.333 | 14.2 |
| TYR | 0.310 | 0.396 | 16.8 | PHE | 0.303 | 0.372 | 16.9 |
| THR | 0.244 | 0.426 | 22.2 | LEU | 0.240 | 0.444 | 22.2 |
| GLT | 0.236 | 0.327 | 21.6 | ARG | 0.200 | 0.219 | 22.8 |
| ALA | 0.196 | 0.197 | 22.7 | TRP | 0.194 | 0.333 | 25.9 |
| PRO | 0.0982 | 0.013 | 15.4 | VAL | ND | ND | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz Calvo, T.; Tejera, N.; McNamara, I.; Langridge, G.C.; Wain, J.; Poolman, M.; Singh, D. Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus epidermidis RP62A. Metabolites 2022, 12, 136. https://doi.org/10.3390/metabo12020136
Díaz Calvo T, Tejera N, McNamara I, Langridge GC, Wain J, Poolman M, Singh D. Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus epidermidis RP62A. Metabolites. 2022; 12(2):136. https://doi.org/10.3390/metabo12020136
Chicago/Turabian StyleDíaz Calvo, Teresa, Noemi Tejera, Iain McNamara, Gemma C. Langridge, John Wain, Mark Poolman, and Dipali Singh. 2022. "Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus epidermidis RP62A" Metabolites 12, no. 2: 136. https://doi.org/10.3390/metabo12020136
APA StyleDíaz Calvo, T., Tejera, N., McNamara, I., Langridge, G. C., Wain, J., Poolman, M., & Singh, D. (2022). Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus epidermidis RP62A. Metabolites, 12(2), 136. https://doi.org/10.3390/metabo12020136