
Plasma Oxylipin Profile Discriminates Ethnicities in Subjects with Non-Alcoholic Steatohepatitis: An Exploratory Analysis

 ,
,  ,
,  ,
,  ,
,
Abstract
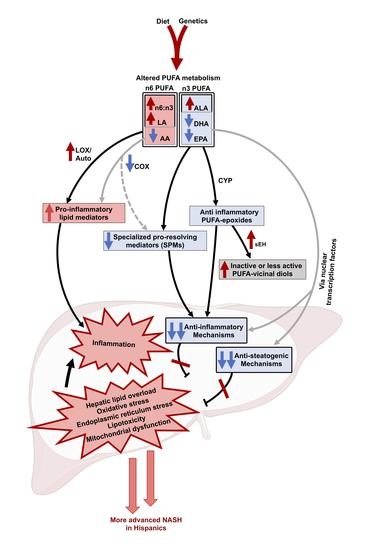
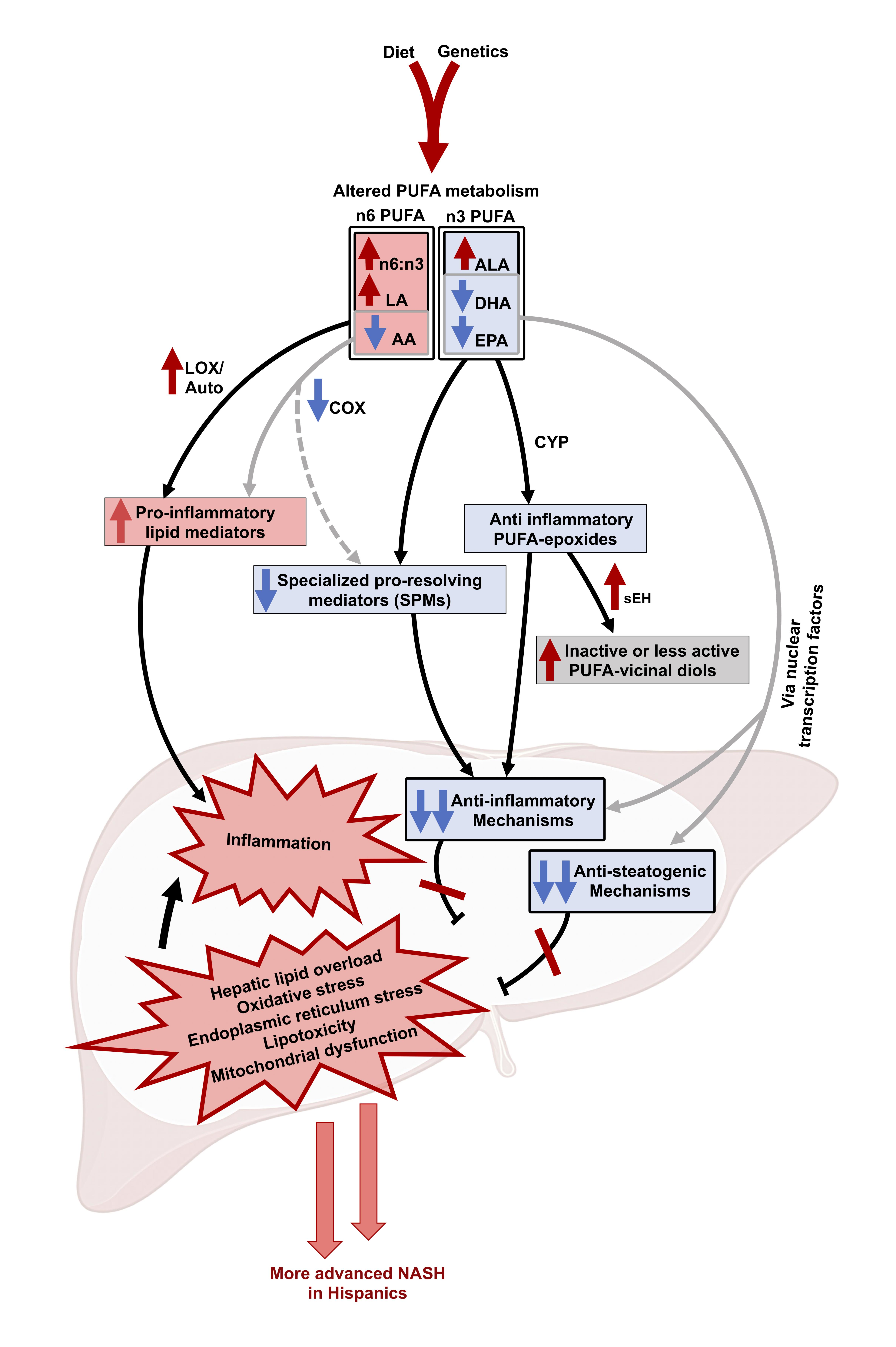
:
1. Introduction
2. Results
2.1. Subject Characteristics
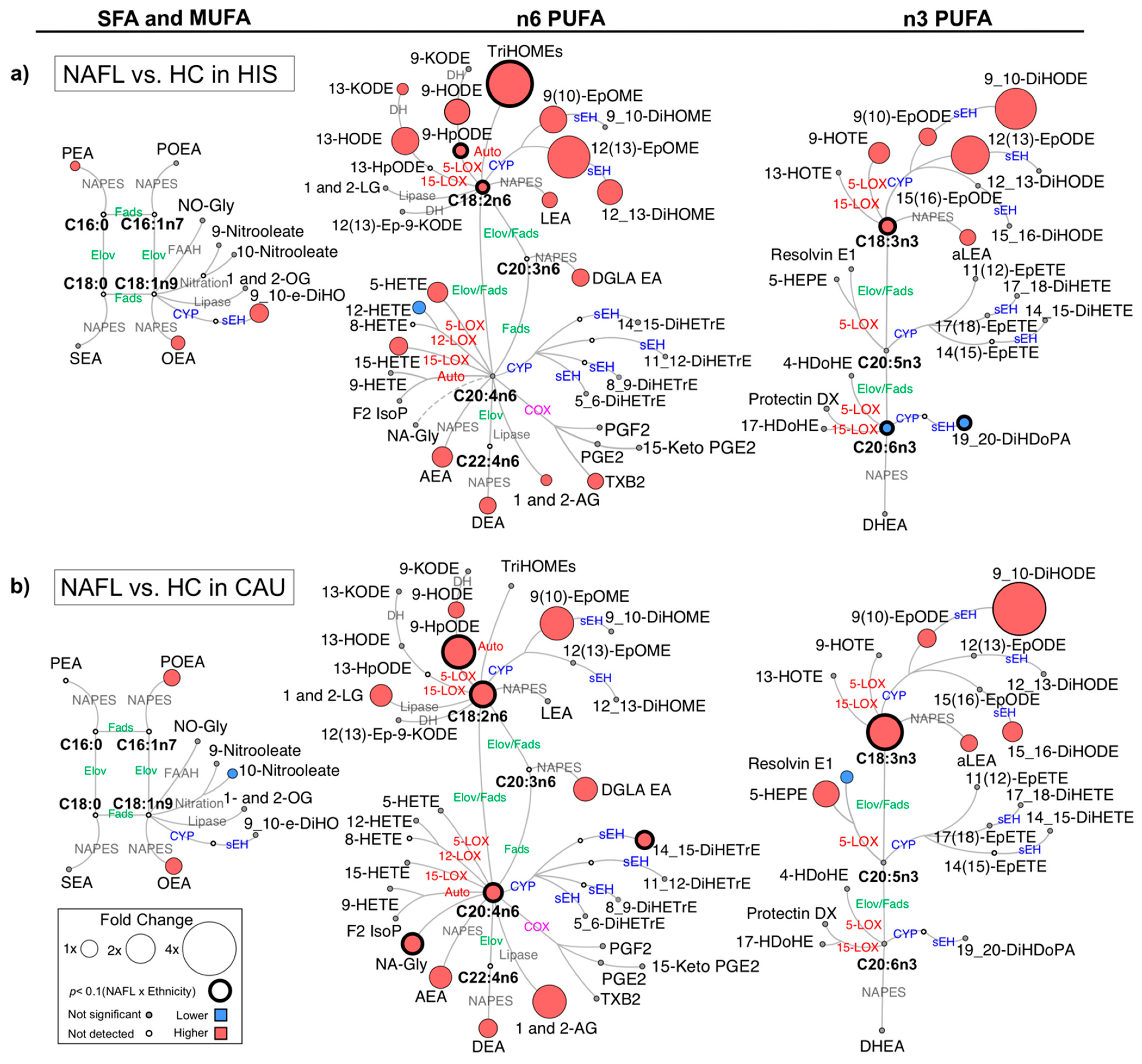
2.2. Ethnicity-Related Alterations in Plasma PUFAs and Lipid Mediator Profiles Characterize NAFL
2.3. Ethnicity-Related Differences in Plasma PUFAs and Lipid Mediators’ Independent of Obesity
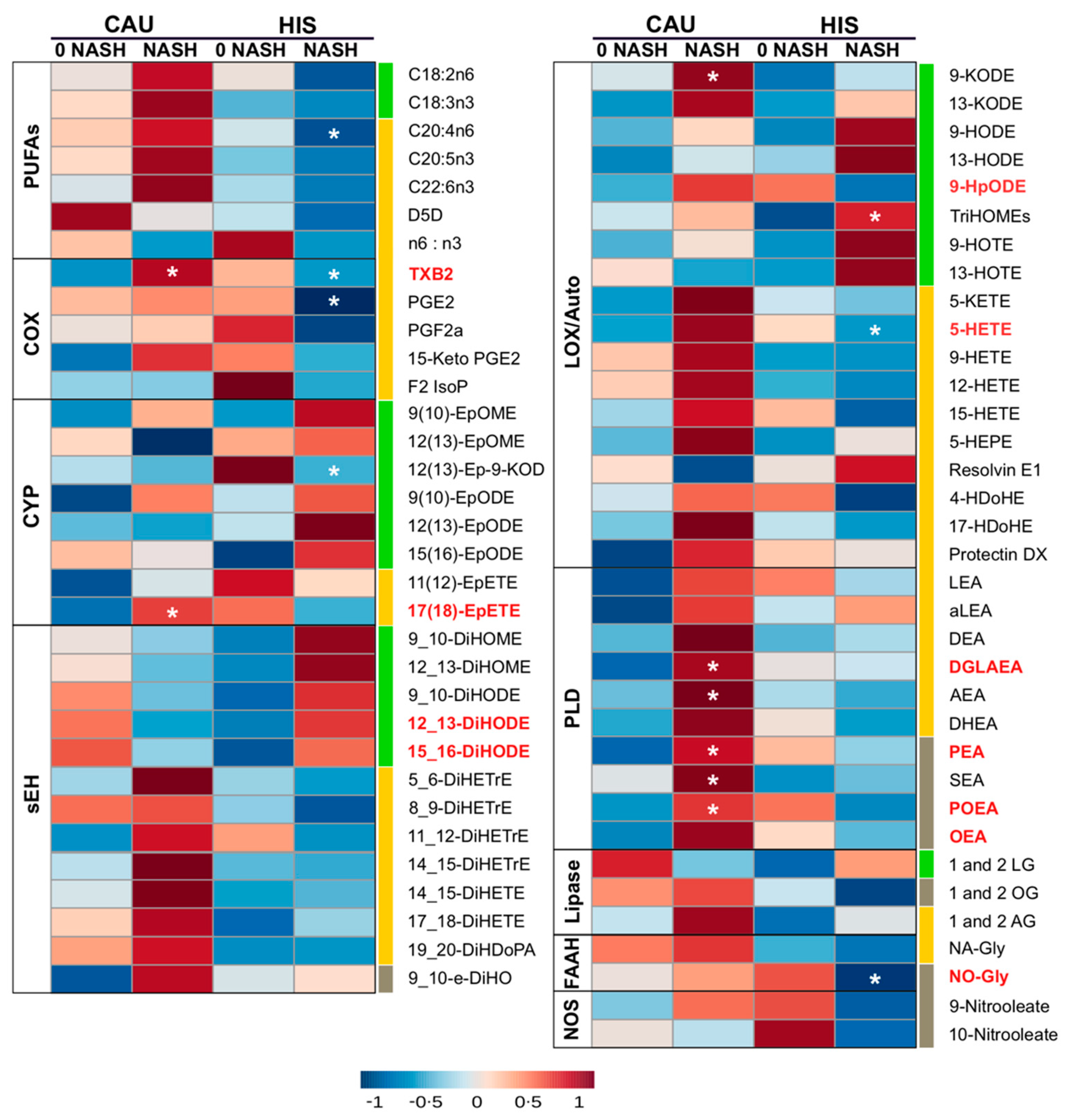
2.4. The Progression to NASH Is Characterized by Ethnicity-Related Alterations in Plasma PUFAs and Lipid Mediator Profiles
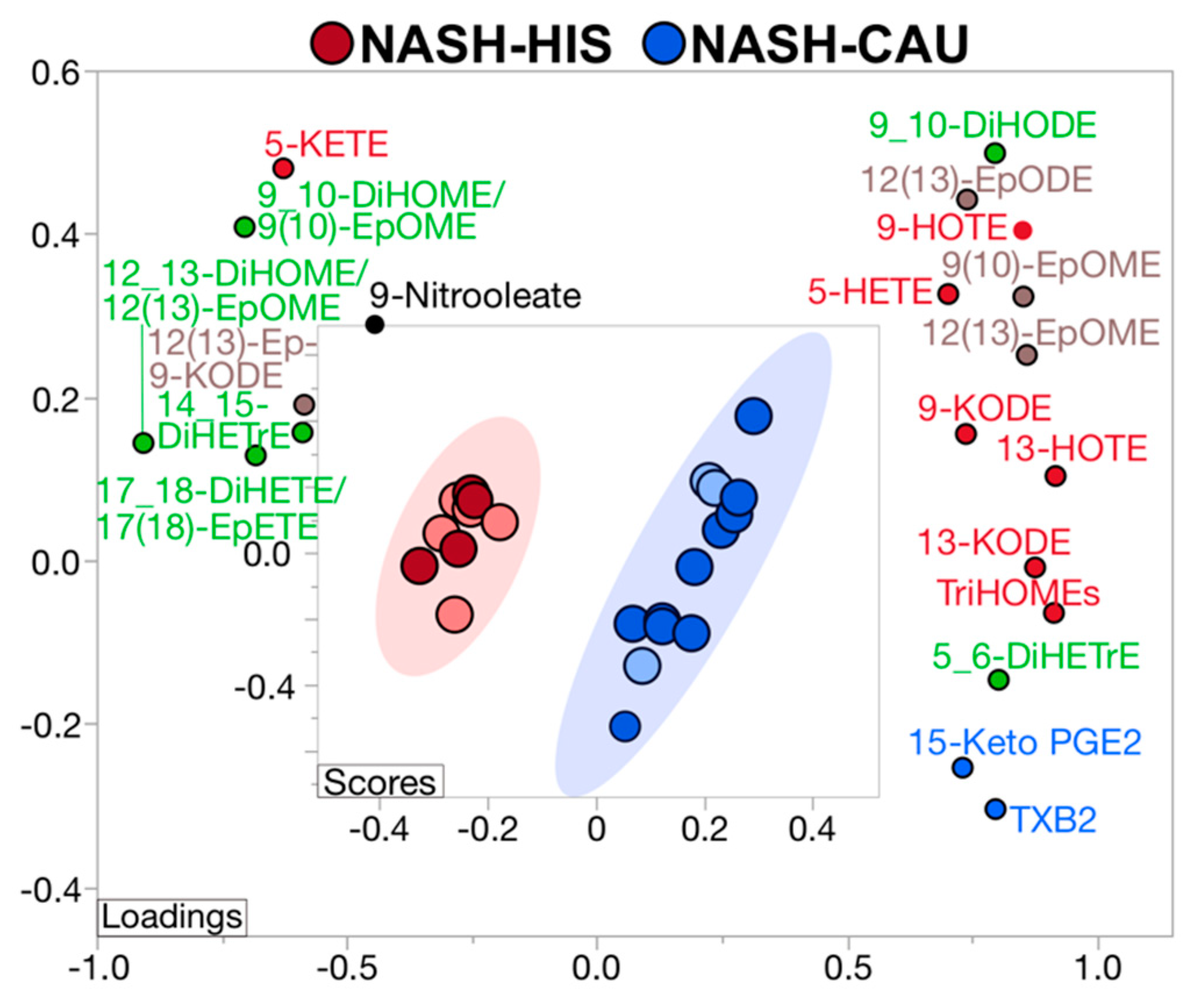
2.5. Plasma OXLs Profile Discriminates between Ethnicities with NASH
3. Discussion
4. Subjects and Methods
4.1. Subjects and Samples
4.2. Plasma Targeted Lipidomic Analysis
4.3. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of Nonalcoholic Fatty Liver Disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiology 2013, 178, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2017, 16, 198–210.e2. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, S.R.; Troy, T.N.; Huo, D.; O’Brien, B.L.; Jensen, D.M.; Hart, J. Influence of ethnicity on histological differences in non-alcoholic fatty liver disease. J. Hepatol. 2009, 50, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. Int. Rev. J. 2015, 6, 513–540. [Google Scholar] [CrossRef] [Green Version]
- Roy, J.; Le Guennec, J.-Y.; Galano, J.-M.; Thireau, J.; Bultel-Poncé, V.; Demion, M.; Oger, C.; Lee, J.C.-Y.; Durand, T. Non-enzymatic cyclic oxygenated metabolites of omega-3 polyunsaturated fatty acid: Bioactive drugs? Biochimie 2016, 120, 56–61. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2016, 31, 1273–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, A.; Teixeira-Clerc, F.; Deveaux, V.; Manin, S.; Lotersztajn, S. The endocannabinoid system as a key mediator during liver diseases: New insights and therapeutic openings. J. Cereb. Blood Flow Metab. 2011, 163, 1432–1440. [Google Scholar] [CrossRef]
- Tam, J.; Liu, J.; Mukhopadhyay, B.; Cinar, R.; Godlewski, G.; Kunos, G. Endocannabinoids in liver disease. Hepatology 2010, 53, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.-C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.-K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [Green Version]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metab. Clin. Exp. 2011, 60, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Walle, P.; Takkunen, M.; Männistö, V.; Vaittinen, M.; Lankinen, M.; Kärjä, V.; Käkelä, P.; Ågren, J.; Tiainen, M.; Schwab, U.; et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism 2016, 65, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, D.; Ossowski, P.; Drozd, A.; Ryterska, K.; Jamioł-Milc, D.; Banaszczak, M.; Kaczorowska, M.; Sabinicz, A.; Raszeja-Wyszomirska, J.; Stachowska, E. Metabolites of arachidonic acid and linoleic acid in early stages of non-alcoholic fatty liver disease—A pilot study. Prostaglandins Other Lipid Mediat. 2015, 121, 184–189. [Google Scholar] [CrossRef]
- Loomba, R.; Quehenberger, O.; Armando, A.; Dennis, E.A. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J. Lipid Res. 2015, 56, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldstein, A.E.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Chung, Y.M.; Berk, M.; Zhang, R.; McIntyre, T.M.; Hazen, S.L. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J. Lipid Res. 2010, 51, 3046–3054. [Google Scholar] [CrossRef] [Green Version]
- Gorden, D.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Kimberly, W.T.; O’Sullivan, J.; Nath, A.K.; Keyes, M.; Shi, X.; Larson, M.G.; Yang, Q.; Long, M.T.; Vasan, R.; Peterson, R.T.; et al. Metabolite profiling identifies anandamide as a biomarker of nonalcoholic steatohepatitis. JCI Insight 2017, 2, 9. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Azar, S.; Nemirovski, A.; Webb, M.; Halpern, Z.; Shibolet, O.; Tam, J. Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity 2016, 25, 94–101. [Google Scholar] [CrossRef]
- Mazi, T.A.; Borkowski, K.; Newman, J.W.; Fiehn, O.; Bowlus, C.L.; Sarkar, S.; Matsukuma, K.; Ali, M.R.; Kieffer, D.A.; Wan, Y.-J.Y.; et al. Ethnicity-specific alterations of plasma and hepatic lipidomic profiles are related to high NAFLD rate and severity in Hispanic Americans, a pilot study. Free Radic. Biol. Med. 2021, 172, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Devarshi, P.P.; Ekimura, S.; Marshall, K.; Mitmesser, S.H. Long-chain omega-3 fatty acid serum concentrations across life stages in the USA: An analysis of NHANES 2011–2012. BMJ Open 2021, 11, e043301. [Google Scholar] [CrossRef]
- Steffen, B.T.; Steffen, L.M.; Tracy, R.; Siscovick, D.; Jacobs, D.; Liu, K.; He, K.; Hanson, N.Q.; Nettleton, J.A.; Tsai, M.Y. Ethnicity, plasma phospholipid fatty acid composition and inflammatory/endothelial activation biomarkers in the Multi-Ethnic Study of Atherosclerosis (MESA). Eur. J. Clin. Nutr. 2012, 66, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.; Mantzioris, E.; Gibson, R.; Ramsden, C.; Muhlhausler, B. The effect of modifying dietary LA and ALA intakes on omega-3 long chain polyunsaturated fatty acid (n-3 LCPUFA) status in human adults: A systematic review and commentary. Prostaglandins Leukot. Essent. Fat. Acids 2015, 95, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Silva, I.; Villalpando, S.; Moreno-Saracho, J.E.; Bernal-Medina, D. Fatty acids intake in the Mexican population. Results of the National Nutrition Survey 2006. Nutr. Metab. 2011, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, L.; Gohlke, H.; Müller-Nurasyid, M.; Heid, I.M.; Palmer, L.; Kompauer, I.; Demmelmair, H.; Illig, T.; Koletzko, B.; Heinrich, J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006, 15, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Lattka, E.; Zeilinger, S.; Illig, T.; Steer, C. Genetic variants of the fatty acid desaturase gene cluster predict amounts of red blood cell docosahexaenoic and other polyunsaturated fatty acids in pregnant women: Findings from the Avon Longitudinal Study of Parents and Children. Am. J. Clin. Nutr. 2010, 93, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Athinarayanan, S.; Jiang, G.; Chalasani, N.; Zhang, M.; Liu, W. Fatty acid desaturase 1 gene polymorphisms control human hepatic lipid composition. Hepatology 2014, 61, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Vernekar, M.; Amarapurkar, D.; Joshi, K.; Singhal, R. Gene polymorphisms of desaturase enzymes of polyunsaturated fatty acid metabolism and adiponutrin and the increased risk of nonalcoholic fatty liver disease. Meta Gene 2017, 11, 152–156. [Google Scholar] [CrossRef]
- Yang, C.; Hallmark, B.; Chai, J.C.; O’Connor, T.D.; Reynolds, L.M.; Wood, A.C.; Seeds, M.; Chen, Y.-D.I.; Steffen, L.M.; Tsai, M.Y.; et al. Impact of Amerind ancestry and FADS genetic variation on omega-3 deficiency and cardiometabolic traits in Hispanic populations. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Li, N.; Chen, M.; Zheng, J.; Qian, Z.; Wang, X.; Huang, C.; Xu, S.; Shi, G. Cyclooxygenase-2 Promotes Hepatocellular Apoptosis by Interacting with TNF-α and IL-6 in the Pathogenesis of Nonalcoholic Steatohepatitis in Rats. Am. J. Dig. Dis. 2013, 58, 2895–2902. [Google Scholar] [CrossRef]
- Du, Y.; Chen, J.; Liu, D.; Bai, Q.; Song, J.; Guan, J.; Gao, J.; Liu, B.; Ma, X. Celecoxib attenuates liver steatosis and inflammation in non-alcoholic steatohepatitis induced by high-fat diet in rats. Mol. Med. Rep. 2011, 4, 811–816. [Google Scholar] [CrossRef]
- Lazić, M.; Inzaugarat, M.E.; Povero, D.; Zhao, I.C.; Chen, M.; Nalbandian, M.; Miller, Y.I.; Cherñavsky, A.C.; Feldstein, A.E.; Sears, D.D. Reduced Dietary Omega-6 to Omega-3 Fatty Acid Ratio and 12/15-Lipoxygenase Deficiency Are Protective against Chronic High Fat Diet-Induced Steatohepatitis. PLoS ONE 2014, 9, e107658. [Google Scholar] [CrossRef] [PubMed]
- López-Parra, M.; Titos, E.; Horrillo, R.; Ferré, N.; González-Périz, A.; Martínez-Clemente, M.; Planagumà, A.; Masferrer, J.; Arroyo, V.; Claria, J. Regulatory effects of arachidonate 5-lipoxygenase on hepatic microsomal TG transfer protein activity and VLDL-triglyceride and apoB secretion in obese mice. J. Lipid Res. 2008, 49, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Rempel, J.D.; Ball, T.B.; Aukema, H.; Minuk, G.Y. Plasma Oxylipins Levels in Nonalcoholic Fatty Liver Disease. Am. J. Dig. Dis. 2020, 65, 3605–3613. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.; Hardesty, J.; Zirnheld, K.; McClain, C.; Warner, D.; Kirpich, I. Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology 2020, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, T.; Djaouti, L.; Demizieux, L.; Gresti, J.; Vergès, B.; Degrace, P. CB1 Antagonism Exerts Specific Molecular Effects on Visceral and Subcutaneous Fat and Reverses Liver Steatosis in Diet-Induced Obese Mice. Diabetes 2010, 59, 926–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei-Hyiaman, U.; Liu, J.; Zhou, L.; Godlewski, G.; Harvey-White, J.; Jeong, W.-I.; Bátkai, S.; Marsicano, G.; Lutz, B.; Buettner, C.; et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J. Clin. Investig. 2008, 118, 3160–3169. [Google Scholar] [CrossRef]
- Weir, N.L.; Nomura, S.O.; Steffen, B.T.; Guan, W.; Karger, A.; Klein, R.; Klein, B.E.; Cotch, M.F.; Tsai, M.Y. Associations between omega-6 polyunsaturated fatty acids, hyperinsulinemia and incident diabetes by race/ethnicity: The Multi-Ethnic Study of Atherosclerosis. Clin. Nutr. 2020, 39, 3031–3041. [Google Scholar] [CrossRef]
- Garg, P.K.; Guan, W.; Nomura, S.; Weir, N.; Karger, A.B.; Duprez, D.; Heckbert, S.R.; Tsai, M.Y. Plasma ω-3 and ω-6 PUFA Concentrations and Risk of Atrial Fibrillation: The Multi-Ethnic Study of Atherosclerosis. J. Nutr. 2021, 151, 1479–1486. [Google Scholar] [CrossRef]
- De Castro, G.S.; Calder, P.C. Non-alcoholic fatty liver disease and its treatment with n-3 polyunsaturated fatty acids. Clin. Nutr. 2018, 37, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; Cassader, M.; Paschetta, E.; Sircana, A. Specialized Proresolving Mediators: Enhancing Nonalcoholic Steatohepatitis and Fibrosis Resolution. Trends Pharmacol. Sci. 2018, 39, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Sayiner, M.; Fazel, Y.; Koenig, A.; Henry, L.; Younossi, Z.M. Current complications and challenges in nonalcoholic steatohepatitis screening and diagnosis. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, A.; Dogru, T.; Ercin, C.; Genc, H.; Celebi, G.; Gurel, H.; Tapan, S.; Cicek, A.; Barcin, C.; Haymana, C.; et al. Betatrophin Levels Are Related to the Early Histological Findings in Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 425. [Google Scholar] [CrossRef]
- Dogru, T.; Kirik, A.; Gurel, H.; Rizvi, A.; Rizzo, M.; Sonmez, A. The Evolving Role of Fetuin-A in Nonalcoholic Fatty Liver Disease: An Overview from Liver to the Heart. Int. J. Mol. Sci. 2021, 22, 6627. [Google Scholar] [CrossRef]
- Vardarajan, B.; Kalia, V.; Manly, J.; Brickman, A.; Reyes-Dumeyer, D.; Lantigua, R.; Ionita-Laza, I.; Jones, D.P.; Miller, G.W.; Mayeux, R. Differences in plasma metabolites related to Alzheimer’s disease, APOE ε4 status, and ethnicity. Alzheimers Dementia Transl. Res. Clin. Interv. 2020, 6, e12025. [Google Scholar] [CrossRef] [PubMed]
- Vantaku, V.; Donepudi, S.R.; Piyarathna, D.W.B.; Amara, C.S.; Ambati, C.S.R.; Tang, W.; Putluri, V.; Chandrashekar, D.S.; Varambally, S.; Terris, M.K.; et al. Large-scale profiling of serum metabolites in African American and European American patients with bladder cancer reveals metabolic pathways associated with patient survival. Cancer 2019, 125, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Valkengoed, I.G.; Argmann, C.; Ghauharali-van der Vlugt, K.; Aerts, J.M.; Brewster, L.M.; Peters, R.; Vaz, F.M.; Houtkooper, R.H. Ethnic differences in metabolite signatures and type 2 diabetes: A nested case–control analysis among people of South Asian, African and European origin. Nutr. Diabetes 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musa-Veloso, K.; Venditti, C.; Lee, H.Y.; Darch, M.; Floyd, S.; West, S.; Simon, R. Systematic review and meta-analysis of controlled intervention studies on the effectiveness of long-chain omega-3 fatty acids in patients with nonalcoholic fatty liver disease. Nutr. Rev. 2018, 76, 581–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Li, S.; Li, J.; Wang, J.; Zhang, R.; Zhou, Y.; Yin, Q.; Zheng, Y.; Wang, F.; Xia, Y.; et al. Effects of Omega-3 Fatty Acid in Nonalcoholic Fatty Liver Disease: A Meta-Analysis. Gastroenterol. Res. Pract. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hammock, B.D.; McReynolds, C.B.; Wagner, K.; Buckpitt, A.; Cortes-Puch, I.; Croston, G.; Lee, K.S.S.; Yang, J.; Schmidt, W.K.; Hwang, S.H. Movement to the Clinic of Soluble Epoxide Hydrolase Inhibitor EC5026 as an Analgesic for Neuropathic Pain and for Use as a Nonaddictive Opioid Alternative. J. Med. Chem. 2021, 64, 1856–1872. [Google Scholar] [CrossRef]
- Lazaar, A.L.; Yang, L.; Boardley, R.L.; Goyal, N.S.; Robertson, J.; Baldwin, S.J.; Newby, D.E.; Wilkinson, I.B.; Tal-Singer, R.; Mayer, R.J.; et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.-J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Pramfalk, C.; Pavlides, M.; Banerjee, R.; McNeil, C.A.; Neubauer, S.; Karpe, F.; Hodson, L. Sex-Specific Differences in Hepatic Fat Oxidation and Synthesis May Explain the Higher Propensity for NAFLD in Men. J. Clin. Endocrinol. Metab. 2015, 100, 4425–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, T.L.; Gray, I.J.; Newman, J.W. Plasma and serum oxylipin, endocannabinoid, bile acid, steroid, fatty acid and nonsteroidal anti-inflammatory drug quantification in a 96-well plate format. Anal. Chim. Acta 2020, 1143, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. Quantitative Trait Loci Analysis Using the False Discovery Rate. Genetics 2005, 171, 783–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Westerhuis, J.A.; Hoefsloot, H.C.; Smit, S.; Vis, D.J.; Smilde, A.K.; Van Velzen, E.J.J.; Van Duijnhoven, J.P.M.; Van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Byrne, T.; Johansson, E.; Trygg, J.; Vikström, C. Multi-and Megavariate Data Analysis Basic Principles and Applications; Umetrics Academy: Umeå, Sweden, 2013; Volume 1. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NAFL-HIS | NAFL-CAU | p-Value * | |
|---|---|---|---|
| Plasma, n (F/M) | 10 (7/3) | 8 (4/4) | - |
| Age (years) | 47 ± 15 | 50 ± 18 | 0.6 |
| DM, yes (%) | 4 (40) | 4 (50) | 1 |
| FBG mmol/L | 101 ± 14 | 94 ± 13 | 0.3 |
| Cholesterol (mg/dL) | 172 ± 28 | 166 ± 34 | 0.7 |
| TG (mg/dL) | 135 ± 79 | 129 ± 70 | 0.8 |
| HDL (mg/dL) | 44 ± 6 | 43 ± 8 | 0.7 |
| LDL (mg/dL) | 107 ± 24 | 99 ± 35 | 0.4 |
| HbA1c (%) | 6 ± 1 | 6 ± 1 | 0.7 |
| AST (U/L) | 31 ± 13 | 26 ± 9 | 0.6 |
| ALT (U/L) | 40 ± 27 | 31 ± 11 | 0.9 |
| Platelet | 280 ± 84 | 302 ± 110 | 0.8 |
| NAS | 3 ± 3 | 3 ± 1 | 0.7 |
| Steatosis (%) | |||
| <5% | 4 (40) | 1 (12.5) | 0.9 |
| 5 to ≤33% | 3 (30) | 5 (62.5) | |
| 34 to ≤66 % | 2 (20) | 2 (25) | |
| >66% | 1 (10.0) | 0 (0) | |
| Inflammation (%) | |||
| None | 3 (30) | 1 (12) | 0.6 |
| Mild | 1 (10) | 4 (50) | |
| Moderate | 4 (40) | 3 (38) | |
| Severe | 2 (20) | 0 (0) | |
| Ballooning (%) | |||
| None | 4 (40) | 5 (62) | 0.3 |
| Few | 5 (50) | 3 (38) | |
| Many | 1 (10) | 0 (0) | |
| Fibrosis (%) | |||
| None | 7 (70) | 7 (88) | 0.3 |
| 1A | 0 (0) | 1 (12) | |
| 2 | 1 (10) | 0 (0) | |
| 4 | 2 (20) | 0 (0) | |
| NASH-HIS | NASH-CAU | p-Value * | |
|---|---|---|---|
| Plasma, n (F/M) | 12 (9/3) | 17 (11/6) | - |
| Age (years) | 49 ± 8 | 50 ± 13 | 0.8 |
| BMI (Kg/m2) | 41 ± 8 | 37 ± 7 | 0.6 |
| NAS | 5 ± 2 | 5 ± 1 | 0.9 |
| Steatosis (%) | |||
| <5% | 2 (17) | 0 (0) | 0.4 |
| 5 to ≤33% | 4 (33) | 7 (41) | |
| 34 to ≤66 % | 3 (25) | 4 (24) | |
| >66% | 3 (25) | 6 (35) | |
| Inflammation (%) | |||
| None | 0 (0) | 0 (0) | 0.1 |
| Mild | 2 (17) | 3 (18) | |
| Moderate | 6 (50) | 13 (76) | |
| Severe | 4 (33) | 1 (6) | |
| Ballooning (%) | |||
| None | 0 (0) | 0 (0) | 0.6 |
| Few | 8 (67) | 13 (76) | |
| Many | 4 (33) | 4 (24) | |
| Fibrosis (%) | |||
| None | 5 (41) | 4 (23) | 0.8 |
| 1a, b, c | 2 (17) | 6 (35) | |
| 2 | 2 (17) | 3 (18) | |
| 3 | 2 (17) | 2 (12) | |
| 4 | 1 (8) | 2 (12) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazi, T.A.; Borkowski, K.; Fiehn, O.; Bowlus, C.L.; Sarkar, S.; Matsukuma, K.; Ali, M.R.; Kieffer, D.A.; Wan, Y.-J.Y.; Stanhope, K.L.; et al. Plasma Oxylipin Profile Discriminates Ethnicities in Subjects with Non-Alcoholic Steatohepatitis: An Exploratory Analysis. Metabolites 2022, 12, 192. https://doi.org/10.3390/metabo12020192
Mazi TA, Borkowski K, Fiehn O, Bowlus CL, Sarkar S, Matsukuma K, Ali MR, Kieffer DA, Wan Y-JY, Stanhope KL, et al. Plasma Oxylipin Profile Discriminates Ethnicities in Subjects with Non-Alcoholic Steatohepatitis: An Exploratory Analysis. Metabolites. 2022; 12(2):192. https://doi.org/10.3390/metabo12020192
Chicago/Turabian StyleMazi, Tagreed A., Kamil Borkowski, Oliver Fiehn, Christopher L. Bowlus, Souvik Sarkar, Karen Matsukuma, Mohamed R. Ali, Dorothy A. Kieffer, Yu-Jui Y. Wan, Kimber L. Stanhope, and et al. 2022. "Plasma Oxylipin Profile Discriminates Ethnicities in Subjects with Non-Alcoholic Steatohepatitis: An Exploratory Analysis" Metabolites 12, no. 2: 192. https://doi.org/10.3390/metabo12020192
APA StyleMazi, T. A., Borkowski, K., Fiehn, O., Bowlus, C. L., Sarkar, S., Matsukuma, K., Ali, M. R., Kieffer, D. A., Wan, Y.-J. Y., Stanhope, K. L., Havel, P. J., Newman, J. W., & Medici, V. (2022). Plasma Oxylipin Profile Discriminates Ethnicities in Subjects with Non-Alcoholic Steatohepatitis: An Exploratory Analysis. Metabolites, 12(2), 192. https://doi.org/10.3390/metabo12020192