
Combined Targeted and Untargeted Profiling of HeLa Cells Deficient in Purine De Novo Synthesis

 , , , , , ,
, , , , , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
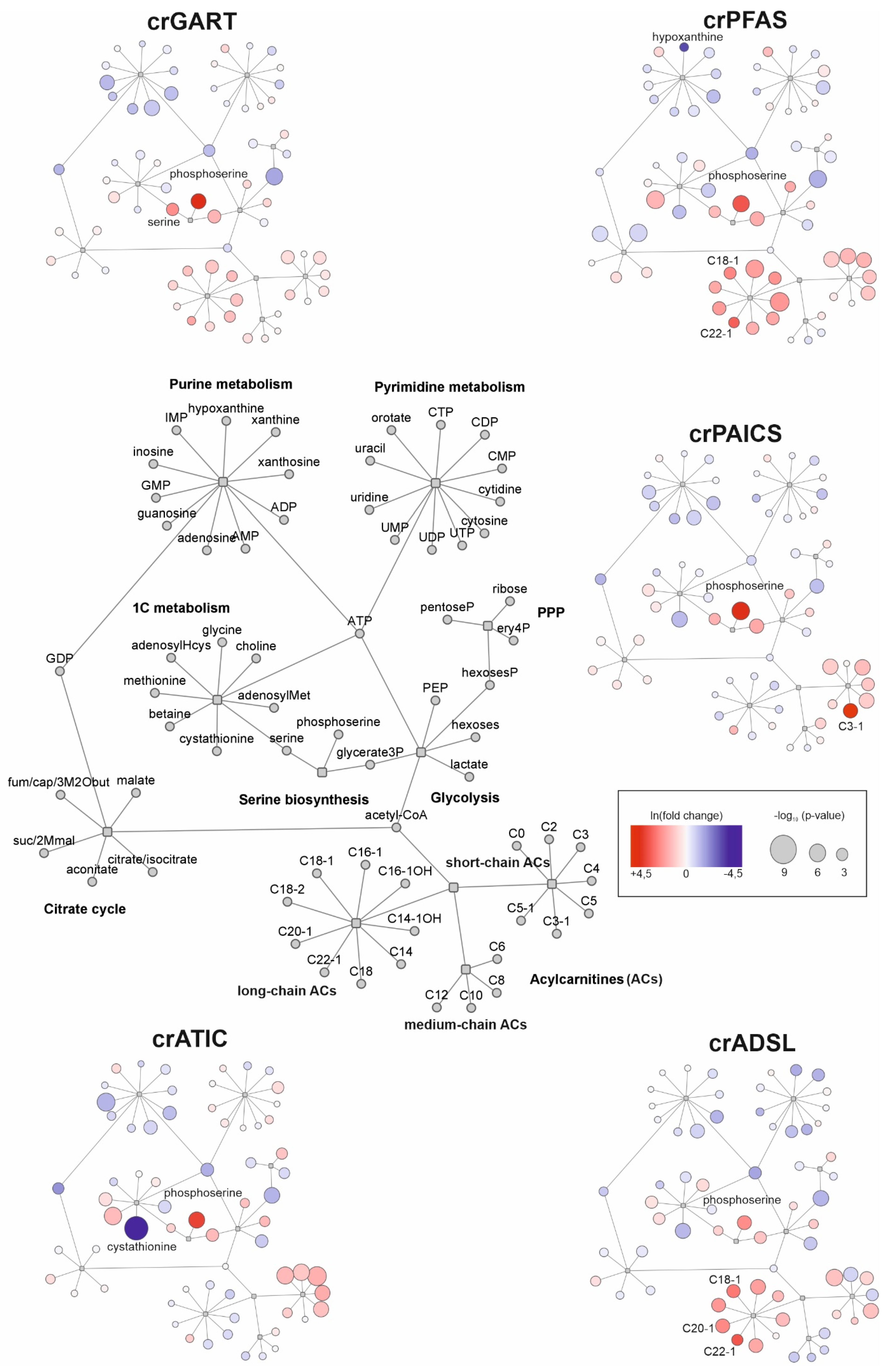
2.1. Untargeted Metabolomic Analysis
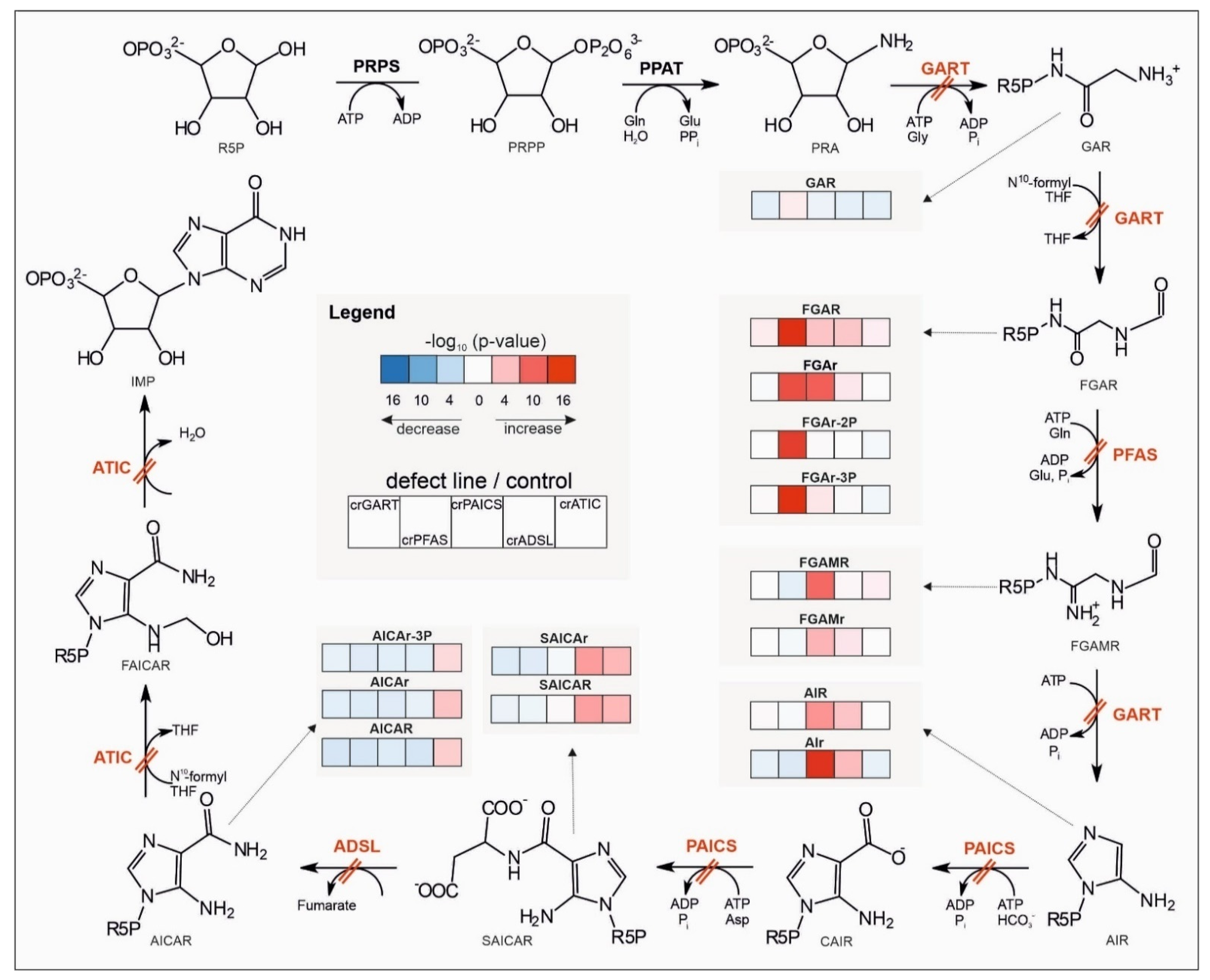
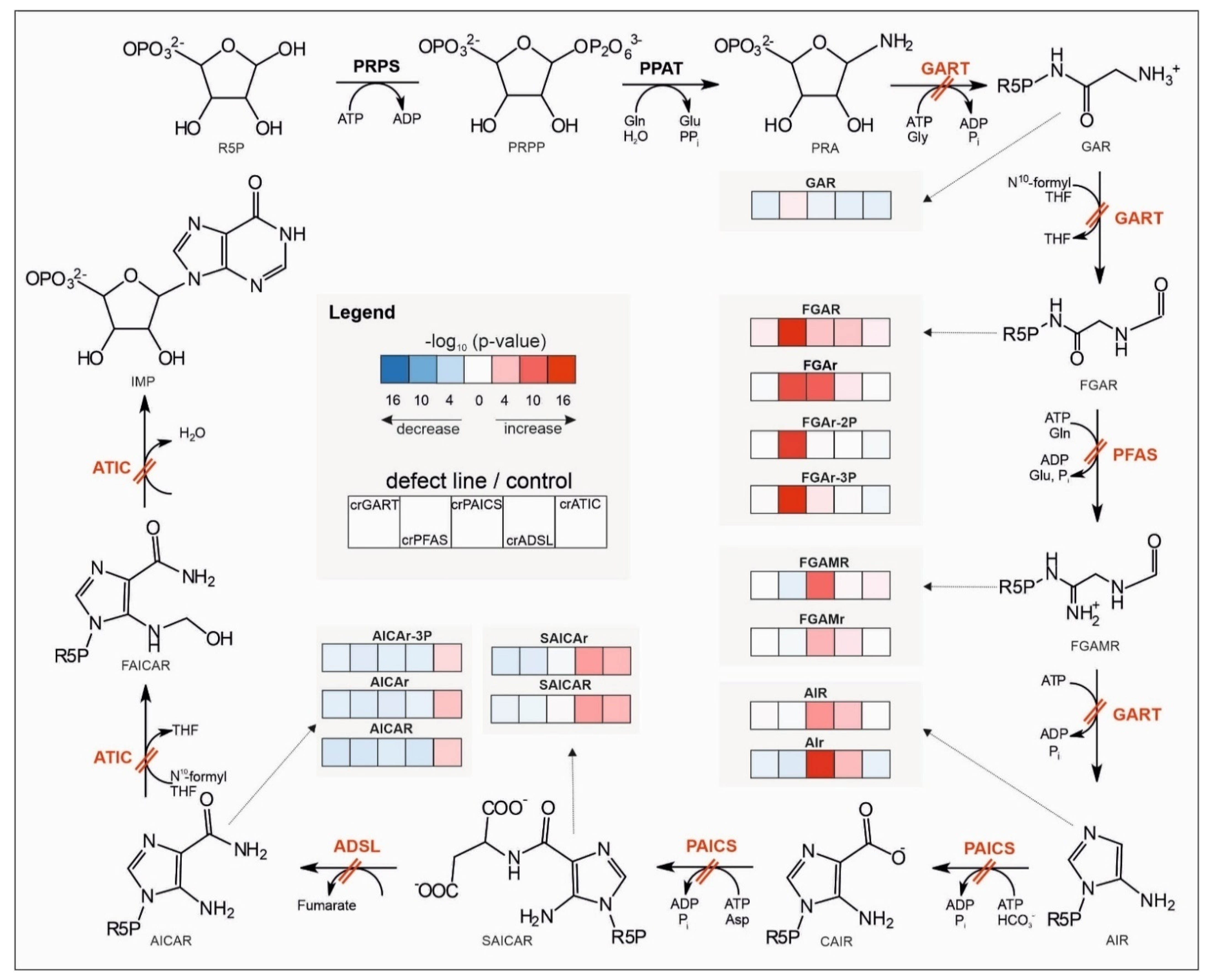
2.2. Targeted Metabolomic Analysis
3. Discussion
3.1. Untargeted Metabolomics
3.2. Targeted Metabolomics
3.2.1. Central Energy Nucleotide Metabolism
3.2.2. Carbohydrate Metabolism
3.2.3. Metabolism of Acylcarnitines
3.2.4. One-Carbon Metabolism
4. Materials and Methods
4.1. Chemicals
4.2. Cell Cultivation, Harvesting, and Sample Preparation
4.3. Untargeted Metabolomic Analysis
4.4. Targeted Metabolomic Analysis
4.5. Data Processing
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Curto, R.; Voit, E.O.; Sorribas, A.; Cascante, M. Mathematical models of purine metabolism in man. Math. Biosci. 1998, 151, 1–49. [Google Scholar] [CrossRef]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible Compartmentalization of de Novo Purine Biosynthetic Complexes in Living Cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Yamaoka, T.; Honda, S.; Miwa, Y.; Katashima, R.; Moritani, M.; Yoshimoto, K.; Hayashi, Y.; Itakura, M. The Rate of Cell and Growth Is Regulated by Purine Biosynthesis via ATP Production G, to S Phase Transition. J. Biochem. 2000, 128, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Gam, J.; French, J.B.; Zhao, H.; An, S.; Benkovic, S.J. Mapping Protein-Protein Proximity in the Purinosome. J. Biol. Chem. 2012, 287, 36201–36207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; Deng, Y.; Tomsho, J.W.; Kyoung, M.; Benkovic, S.J. Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 12872–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, J.B.; Jones, S.A.; Deng, H.; Pedley, A.M.; Kim, D.; Chan, C.Y.; Hu, H.; Pugh, R.J.; Zhao, H.; Zhang, Y.; et al. Spatial colocalization and functional link of purinosomes with mitochondria. Science 2016, 351, 733–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareek, V.; Tian, H.; Winograd, N.; Benkovic, S.J. Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 2020, 368, 283–290. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yano, M.; Kondo, M.; Sasaki, H.; Hino, S.; Katashima, R.; Moritani, M.; Itakura, M. Feedback Inhibition of Amidophosphoribosyltransferase Regulates the Rate of Cell Growth via Purine Nucleotide, DNA, and Protein Syntheses. J. Biol. Chem. 2001, 276, 21285–21291. [Google Scholar] [CrossRef] [Green Version]
- Vergis, J.M.; Bulock, K.G.; Fleming, K.G.; Beardsley, G. Human 5-Aminoimidazole-4-carboxamide Ribonucleotide Transformylase/Inosine 5′-Monophosphate Cyclohydrolase. A bifunctional protein requiring dimerization for transformylase activity but not for cyclohydrolase activity. J. Biol. Chem. 2001, 276, 7727–7733. [Google Scholar] [CrossRef] [Green Version]
- French, J.B.; Zhao, H.; An, S.; Niessen, S.; Deng, Y.; Cravatt, B.F.; Benkovic, S.J. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. USA 2013, 110, 2528–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.; Kyoung, M.; Allen, J.J.; Shokat, K.M.; Benkovic, S.J. Dynamic Regulation of a Metabolic Multi-enzyme Complex by Protein Kinase CK2. J. Biol. Chem. 2010, 285, 11093–11099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Fridman, A.; Blackledge, W.; Connely, S.; Wilson, I.A.; Pilz, R.; Boss, G.R. The Phosphatidylinositol 3-Kinase/Akt Cassette Regulates Purine Nucleotide Synthesis. J. Biol. Chem. 2009, 284, 3521–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramond, F.; Rio, M.; Héron, B.; Imbard, A.; Marie, S.; Billiemaz, K.; Denommé-Pichon, A.; Kuentz, P.; Ceballos, I.; Piraud, M.; et al. AICA -ribosiduria due to ATIC deficiency: Delineation of the phenotype with three novel cases, and long-term update on the first case. J. Inherit. Metab. Dis. 2020, 43, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Marie, S.; Heron, B.; Bitoun, P.; Timmerman, T.; Van Den Berghe, G.; Vincent, M.-F. AICA-Ribosiduria: A Novel, Neurologically Devastating Inborn Error of Purine Biosynthesis Caused by Mutation of ATIC. Am. J. Hum. Genet. 2004, 74, 1276–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baresova, V.; Škopová, V.; Sikora, J.; Patterson, D.; Sovova, J.; Zikanova, M.; Kmoch, S. Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum. Mol. Genet. 2012, 21, 1534–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeken, J.; Wadman, S.K.; Duran, M.; Van Sprang, F.J.; Beemer, F.A.; Holl, R.A.; Theunissen, P.M.; De Cock, P.; van den Bergh, F.; Vincent, M.F.; et al. Adenylosuccinase deficiency: An inborn error of purine nucleotide synthesis. Eur. J. Pediatr. 1988, 148, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymanska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelet, A.; Skopova, V.; Steuerwald, U.; Baresova, V.; Zarhrate, M.; Plaza, J.-M.; Hnizda, A.; Krijt, M.; Souckova, O.; Wibrand, F.; et al. PAICS deficiency, a new defect of of de nvo purine synthesis resulting in multiple congenital anomalies and fatal outcome. Hum. Mol. Genet. 2019, 28, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A. Inborn errors of purine and pyrimidine metabolism. J. Inherit. Metab. Dis. 2009, 32, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Mazzarino, R.C.; Baresova, V.; Zikánová, M.; Duval, N.; Wilkinson, T.G.; Patterson, D.; Vacano, G.N. The CRISPR-Cas9 crGART HeLa transcriptome: A novel cell model of de novo purine synthesis deficiency. BioRxiv 2020. [CrossRef]
- Mazzarino, R.C.; Baresova, V.; Zikánová, M.; Duval, N.; Wilkinson, T.; Patterson, D.; Vacano, G.N. The CRISPR-Cas9 crADSL HeLa transcriptome: A first step in establishing a model for ADSL deficiency and SAICAR accumulation. Mol. Genet. Metab. Rep. 2019, 21, 100512. [Google Scholar] [CrossRef] [PubMed]
- Mazzarino, R.C.; Baresova, V.; Zikánová, M.; Duval, N.; Wilkinson, T.G.; Patterson, D.; Vacano, G.N. The CRISPR-Cas9 crATIC HeLa transcriptome: Characterization of a novel cellular model of ATIC deficiency and ZMP accumulation. Mol. Genet. Metab. Rep. 2020, 25, 100642. [Google Scholar] [CrossRef] [PubMed]
- Mádrová, L.; Krijt, M.; Barešová, V.; Václavík, J.; Friedecký, D.; Dobešová, D.; Součková, O.; Škopová, V.; Adam, T.; Zikánová, M. Mass spectrometric analysis of purine de novo biosynthesis intermediates. PLoS ONE 2018, 13, e0208947. [Google Scholar] [CrossRef] [PubMed]
- Kouřil, Š.; de Sousa, J.; Václavík, J.; Friedecký, D.; Adam, T. CROP: Correlation-based reduction of feature multiplicities in untargeted metabolomic data. Bioinformatics 2020, 36, 2941–2942. [Google Scholar] [CrossRef] [PubMed]
- Schendel, F.J.; Cheng, Y.S.; Otvos, J.D.; Wehrli, S.; Stubbe, J. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry 1988, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Baresova, V.; Krijt, M.; Skopova, V.; Součková, O.; Kmoch, S.; Zikanova, M. CRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation. Mol. Genet. Metab. 2016, 119, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Zikanova, M.; Tylki-Szymanska, A.; Krijt, J.; Bogdanska, A.; Gradowska, W.; Mullerova, K.; Sykut-Cegielska, J.; Kmoch, S.; Pronicka, E. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Mol. Genet. Metab. 2008, 94, 435–442. [Google Scholar] [CrossRef]
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant adsl enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Sabina, R.L.; Holmes, E.W.; Becker, M.A. The Enzymatic Synthesis of 5-Amino-4-Imidazolecarboxamide Riboside Triphosphate (ZTP). Science 1984, 223, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Daignan-Fornier, B.; Pinson, B. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5’-Monophosphate (AICAR), a Highly Conserved Purine Intermediate with Multiple Effects. Metabolites 2012, 2, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; Simmonds, H.A. Purines: Basic and Clinical Aspects; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1991. [Google Scholar] [CrossRef]
- van den Bergh, F.; Vincent, M.F.; Jaeken, J.; van den Bergh, G. Residual adenylosuccinase activities in fibroblasts of adenylosuccinase-deficient children: Parallel deficiency with adenylosuccinate and succinyl-AICAR in profoundly retarded patients and non-parallel deficiency in a mildly retarded girl. J. Inherit. Metab. Dis. 1993, 16, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.E.; Walton, G.M. Adenosine Triphosphate Conservation in Metabolic Regulation. J. Biol. Chem. 1967, 242, 3239–3241. [Google Scholar] [CrossRef]
- Shen, L.; Fall, L.; Walton, G.M.; Atkinson, D.E. Interaction between energy charge and metabolite modulation in the regulation of enzymes of amphibolic sequences. Phosphofructokinase and pyruvate dehydrogenase. Biochemistry 1968, 7, 4041–4045. [Google Scholar] [CrossRef] [PubMed]
- Swedes, J.S.; Sedo, R.J.; E Atkinson, D. Relation of growth and protein synthesis to the adenylate energy charge in an adenine-requiring mutant of Escherichia coli. J. Biol. Chem. 1975, 250, 6930–6938. [Google Scholar] [CrossRef]
- De La Fuente, I.M.; Cortes, J.M.; Valero, E.; Desroches, M.; Rodrigues, S.; Malaina, I.; Martínez, L. On the Dynamics of the Adenylate Energy System: Homeorhesis vs. Homeostasis. PLoS ONE 2014, 9, e108676. [Google Scholar] [CrossRef] [Green Version]
- Boutchueng-Djidjou, M.; Collard-Simard, G.; Fortier, S.; Hébert, S.; Kelly, I.; Landry, C.R.; Faure, R.L. The Last Enzyme of the De Novo Purine Synthesis Pathway 5-aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase/IMP Cyclohydrolase (ATIC) Plays a Central Role in Insulin Signaling and the Golgi/Endosomes Protein Network. Mol. Cell. Proteom. 2015, 14, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Kit, S. The biosynthesis of free glycine and serine by tumors. Cancer Res. 1955, 15, 715–718. [Google Scholar] [PubMed]
- Tedeschi, P.M.; Markert, E.K.; Gounder, M.; Lin, H.; Dvorzhinski, D.; Dolfi, S.C.; Chan, L.L.-Y.; Qiu, J.; DiPaola, R.S.; Hirshfield, K.M.; et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013, 4, e877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zogg, C.K. Phosphoglycerate Dehydrogenase: Potential Therapeutic Target and Putative Metabolic Oncogene. J. Oncol. 2014, 2014, 524101. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martín, D.; Martinez-Martin, N.; Blas-García, A.; Morales, J.M.; Martí-Cabrera, M.; Monleon, D.; Apostolova, N. Metabolomics of the effect of AMPK activation by AICAR on human umbilical vein endothelial cells. Int. J. Mol. Med. 2012, 29, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, D.; Jiang, Y. Function, Detection and Alteration of Acylcarnitine Metabolism in Hepatocellular Carcinoma. Metabolites 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, E.S.; Tan, M.L.S.; Stevens, R.D.; Low, Y.L.; Muehlbauer, M.J.; Goh, D.L.M.; Ilkayeva, O.R.; Wenner, B.R.; Bain, J.R.; Lee, J.J.M.; et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia 2010, 53, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-Carboxamide Ribonucleoside. A Specific Method for Activating AMP-Activated Protein Kinase in Intact Cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.R.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Donti, T.R.; Cappuccio, G.; Hubert, L.; Neira, J.; Atwal, P.S.; Miller, M.J.; Cardon, A.L.; Sutton, V.R.; Porter, B.E.; Baumer, F.; et al. Diagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum. Mol. Genet. Metab. Rep. 2016, 8, 61–66. [Google Scholar] [CrossRef]
- Subedi, A.; Muroi, M.; Futamura, Y.; Kawamura, T.; Aono, H.; Nishi, M.; Ryo, A.; Watanabe, N.; Osada, H. A novel inhibitor of tumorspheres reveals the activation of the serine biosynthetic pathway upon mitochondrial inhibition. FEBS Lett. 2019, 593, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Labuschagne, C.F.; Van Den Broek, N.J.F.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D.K. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. S-Adenosylmethionine. Int. J. Biochem. Cell Biol. 2000, 32, 391–395. [Google Scholar] [CrossRef]
- Kim, P.B.; Nelson, J.W.; Breaker, R.R. An Ancient Riboswitch Class in Bacteria Regulates Purine Biosynthesis and One-Carbon Metabolism. Mol. Cell 2015, 57, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.; Rabinowitz, J.D. ZMP: A Master Regulator of One-Carbon Metabolism. Mol. Cell 2015, 57, 203–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtowicz, P.; Zrostlíková, J.; Veronika, Š.; Dostálová, E.; Žídková, L.; Bruheim, P.; Adam, T. Comprehensive Two-Dimensional Gas Chromatography Coupled to Time-of-Flight Mass Spectrometry in Human Metabolomics. In Gas Chromatography; IntechOpen: London, UK, 2008; pp. 29–50. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlíková, R.; Široká, J.; Friedecký, D.; Faber, E.; Hrdá, M.; Mičová, K.; Fikarová, I.; Gardlo, A.; Janečková, H.; Vrobel, I.; et al. Metabolite Profiling of the Plasma and Leukocytes of Chronic Myeloid Leukemia Patients. J. Proteome Res. 2016, 15, 3158–3166. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, W.S. Robust Locally Weighted Regression and Smoothing Scatterplots. J. Am. Stat. Assoc. 1979, 74, 829–836. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Pawlowsky-Glahn, V.; Egozcue, J.J.; Tolosana-Delgado, R. Modelling and Analysis of Compositional Data; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mádrová, L.; Součková, O.; Brumarová, R.; Dobešová, D.; Václavík, J.; Kouřil, Š.; de Sousa, J.; Friedecká, J.; Friedecký, D.; Barešová, V.; et al. Combined Targeted and Untargeted Profiling of HeLa Cells Deficient in Purine De Novo Synthesis. Metabolites 2022, 12, 241. https://doi.org/10.3390/metabo12030241
Mádrová L, Součková O, Brumarová R, Dobešová D, Václavík J, Kouřil Š, de Sousa J, Friedecká J, Friedecký D, Barešová V, et al. Combined Targeted and Untargeted Profiling of HeLa Cells Deficient in Purine De Novo Synthesis. Metabolites. 2022; 12(3):241. https://doi.org/10.3390/metabo12030241
Chicago/Turabian StyleMádrová, Lucie, Olga Součková, Radana Brumarová, Dana Dobešová, Jan Václavík, Štěpán Kouřil, Julie de Sousa, Jaroslava Friedecká, David Friedecký, Veronika Barešová, and et al. 2022. "Combined Targeted and Untargeted Profiling of HeLa Cells Deficient in Purine De Novo Synthesis" Metabolites 12, no. 3: 241. https://doi.org/10.3390/metabo12030241
APA StyleMádrová, L., Součková, O., Brumarová, R., Dobešová, D., Václavík, J., Kouřil, Š., de Sousa, J., Friedecká, J., Friedecký, D., Barešová, V., Zikánová, M., & Adam, T. (2022). Combined Targeted and Untargeted Profiling of HeLa Cells Deficient in Purine De Novo Synthesis. Metabolites, 12(3), 241. https://doi.org/10.3390/metabo12030241