
Understanding Inborn Errors of Metabolism through Metabolomics


Abstract
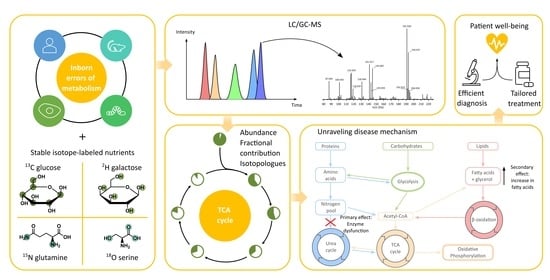
:
1. Introduction
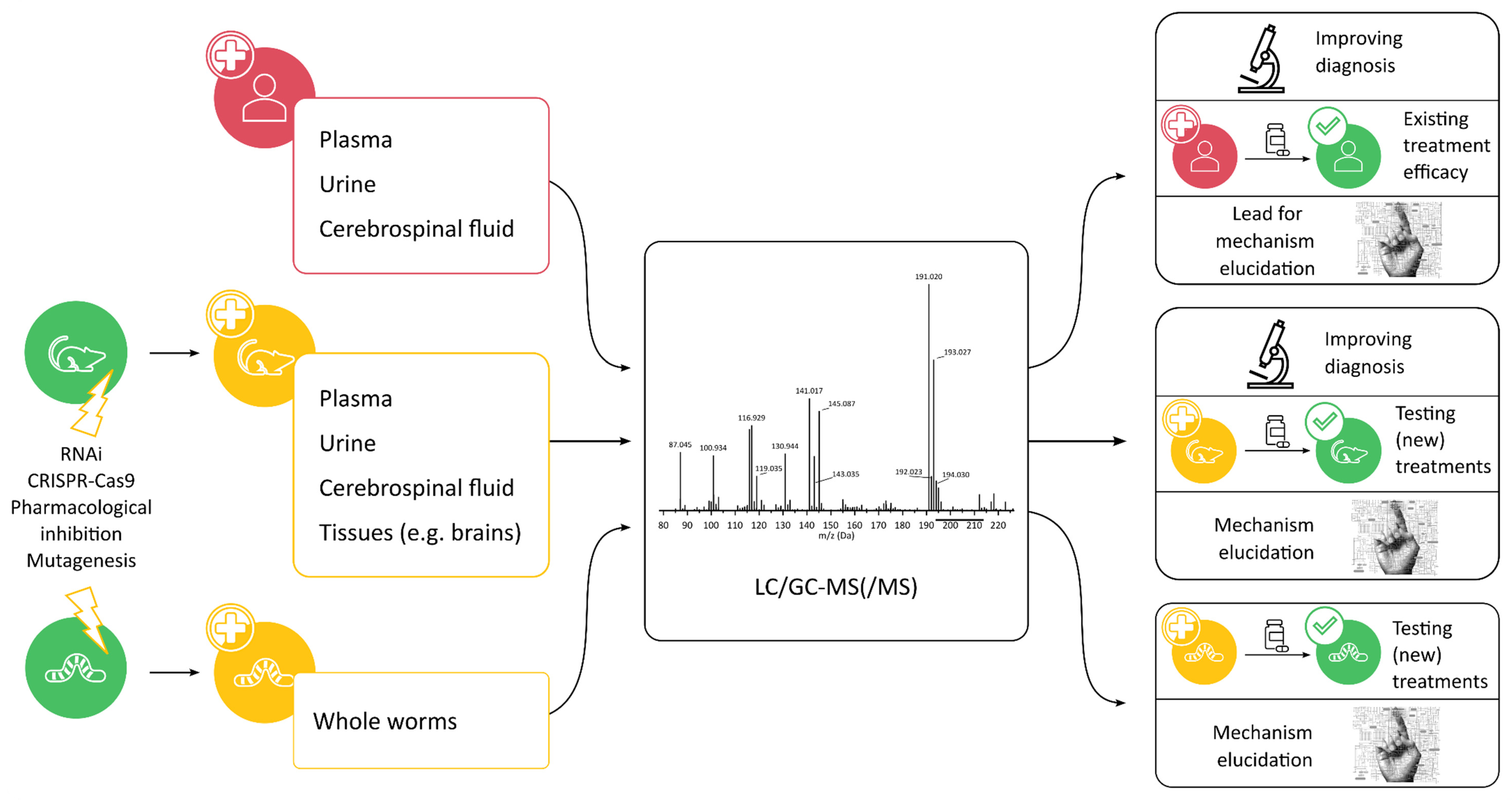
2. In Vivo and Ex Vivo Metabolomics
2.1. Analysis of Patient Samples
2.1.1. Biomarker Discovery for Diagnosis
2.1.2. Treatment Efficacy
2.1.3. Mechanism Elucidation
2.2. Model Organisms to Study IEMs
2.2.1. Mice (Mus musculus)
2.2.2. Worms (Caenorhabditis elegans)
3. In Vitro Metabolomics
3.1. Modified Cell Lines

3.2. Patient Fibroblasts
4. Conclusions and Recommendations
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Inborn Error of Metabolism | Model System | Induced Mutation | Treatment | Stable Isotopes | Key Findings (Affected Pathways/Metabolites) |
|---|---|---|---|---|---|
| Disorders affecting small molecules | |||||
| Phenylketonuria [29] | Human plasma and urine | N/A | Amino acid -medical food (AA-MF) vs. Glycomacro-peptide-MF (GMP-MF) | N/A |
|
| Phenylketonuria [44] | Mouse brains | Chemical ENU induced mutation to Pah enu2+/− mice that were interbred | N/A | N/A |
|
| Alkaptonuria [47] | Mouse and human urine | Inducible knockout of HGD | Nitisinone | 13C6-homogen-tisic acid |
|
| Urea cycle disorders [32] | Human plasma | N/A | Standard of care | N/A |
|
| Urea cycle disorders [36] (CPS1 deficiency) | Human plasma, urine, and breath | N/A | N-carbamyl-L-glutamate | 1-13C-acetate |
|
| Urea cycle disorders [37] (NAGS deficiency) | Human plasma, urine, and breath | N/A | N-carbamyl-L-glutamate | 1-13C-acetate |
|
| Disorders affecting molecule transport | |||||
| GLUT1 deficiency [28] | Human plasma, urine, and cerebrospinal fluid | N/A | Ketogenic diet | N/A |
|
| GLUT1 deficiency [45] | Mouse forebrains | Antisense-GLUT1 injection in embryos | N/A | N/A |
|
| Disorders affecting energy metabolism | |||||
| Fatty acid oxidation disorder [34] (VLCAD deficiency) | Human plasma | N/A | Standard of care vs. triheptanoin | N/A |
|
| LIPT1 deficiency [62] | Human plasma and fibroblasts, mouse model | Transfection with wild-type LIPT1 | N/A | U-13C-glutamine |
|
| EARS2 deficiency [63] | Human fibroblasts | N/A | N/A | N/A |
|
| Mitochondrial dysfunction [59] (SLC25A1 deficiency) | Human fibroblasts | N/A | N/A | U-13C-glucose |
|
| Mitochondrial dysfunction [60] (SLC25A1 deficiency) | Human fibroblasts | Transfection with wild-type SLC25A1 | N/A | N/A |
|
| Mitochondrial dysfunction [50] (Complex I, II, III, and CoQ) | Caenorhabditis elegans | Purchased with missense mutation | N/A | N/A |
|
| Mitochondrial dysfunction [51] (Complex I, II, III, and CoQ, TCA cycle, pyruvate metabolism defect, nutrient-sensing) | Caenorhabditis elegans | Purchased with missense mutation | N/A | 1,6-13C-glucose U-13C-glucose |
|
| Mitochondrial dysfunction [49] (Propionic acidemia) | Caenorhabditis elegans | Purchased with missense mutation | N/A | 1,6-13C2-glucose |
|
| Mitochondrial dysfunction [35] (MELAS) | Human plasma | N/A | Arginine vs. citrulline supplementation | Yes |
|
| Mitochondrial dysfunction [56] (Impaired mtDNA replication) | HEK-293-derived cells | Transfection with mutant DNA polymerase gamma to halt mtDNA replication | N/A | 13C-glucose |
|
| Mitochondrial dysfunction [57] (ATP synthase mutation) | Isogenic cell lines from female human osteosarcoma | Mitochondrially targeted zinc-finger nucleases, varying levels of mtDNA mutation | N/A | U-13C-glucose 4-2H-glucose U-13C-glutamine 1-13C-glutamine U-13C-aspartate |
|
| Disorders affecting large molecules | |||||
| Glycogen storage disease [61] (GSD I and III) | Human fibroblasts | N/A | N/A | N/A |
|
| Glycogen storage disease [33] (GSD I) | Human plasma | N/A | N/A | N/A |
|
| Glycogen storage disease [41] (GSD Ia) | Human plasma | N/A | N/A | 1-13C-acetate |
|
| Glycogen storage disease [46] (GSD Ia) | Mice, mouse hepatocytes | Mice expressing inducible CRE-ERT2 recombinase were treated with tamoxifen to delete exon 3 from G6pc | N/A | 1-13C-galactose 2H-galactose |
|
| Lysosomal storage disease [38] (MPS I) | Human urine | N/A | N/A | N/A |
|
| Lysosomal storage disease [39] (MPS III) | Human urine | N/A | N/A | N/A |
|
| Lysosomal storage disease [40] (MPS VI) | Human urine | N/A | N/A | N/A |
|
| Lysosomal storage disease [43] (MPS IIIB) | Mouse plasma | Induced mutation within exon 6 by replacing fragment with cassette | rAAV gene delivery (enzyme replacement therapy) | N/A |
|
| Congenital disorders of glycosylation [64] (PGM1-CDG) | Human fibroblasts | N/A | Galactose | U-13C-glucose U-13C- galactose |
|
| Congenital disorders of glycosylation [65] (MPI-CDG) | Human fibroblasts | N/A | N/A | 1,2-13C-glucose 4-13C-mannose |
|
| Congenital disorders of glycosylation [67] (TRAPPC9 deficiency) | Human fibroblasts | N/A | N/A | U-13C-glucose |
|
| PGM2L1 deficiency [66] | Human fibroblasts | N/A | N/A | N/A |
|
References
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass Spectrometry-Based Metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The Apogee of the Omics Trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Chen, L.; Rabinowitz, J.D. Metabolomics and Isotope Tracing. Cell 2018, 173, 822–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.M.; Burlingame, A.L.; Cronholm, T.; Sjövall, J. Deuterium and Carbon-13 Tracer Studies of Ethanol Metabolism in the Rat by 2H, 1H-Decoupled 13C Nuclear Magnetic Resonance. Biochem. Biophys. Res. Commun. 1974, 56, 828–835. [Google Scholar] [CrossRef]
- Wishart, D.S. NMR Metabolomics: A Look Ahead. J. Magn. Reson. 2019, 306, 155–161. [Google Scholar] [CrossRef]
- Cui, L.; Lu, H.; Lee, Y.H. Challenges and Emergent Solutions for LC-MS/MS Based Untargeted Metabolomics in Diseases. Mass Spectrom. Rev. 2018, 37, 772–792. [Google Scholar] [CrossRef]
- Liang, L.; Sun, F.; Wang, H.; Hu, Z. Metabolomics, Metabolic Flux Analysis and Cancer Pharmacology. Pharmacol. Ther. 2021, 224, 107827. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond Biomarkers and towards Mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Zamboni, N.; Saghatelian, A.; Patti, G.J. Defining the Metabolome: Size, Flux, and Regulation. Mol. Cell 2015, 58, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted Metabolomics. Curr. Protoc. Mol. Biol. 2012, 98, 30.2.1–30.2.24. [Google Scholar] [CrossRef]
- Reijngoud, D.-J. Flux Analysis of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 2018, 41, 309–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.R.; van Karnebeek, C.D.M. Chapter 22—Inborn Errors of Metabolism. In Handbook of Clinical Neurology; Neonatal Neurology; de Vries, L.S., Glass, H.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 162, pp. 449–481. [Google Scholar]
- Bharadwaj, A.; Wahi, N.; Saxena, A. Occurrence of Inborn Errors of Metabolism in Newborns, Diagnosis and Prophylaxis. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 592–616. [Google Scholar] [CrossRef] [PubMed]
- Garrod, A.E. The Croonian Lectures on Inborn Errors of Metabolism. Lancet 1908, 172, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J.; ICIMD Advisory Group; Abdenur, J.; Ali, H.; Artuch, R.; Ballabio, A.; Barshop, B.; et al. An International Classification of Inherited Metabolic Disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef]
- Waters, D.; Adeloye, D.; Woolham, D.; Wastnedge, E.; Patel, S.; Rudan, I. Global Birth Prevalence and Mortality from Inborn Errors of Metabolism: A Systematic Analysis of the Evidence. J. Glob. Health 2018, 8, 021102. [Google Scholar] [CrossRef]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The Incidence of Inherited Metabolic Disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Saudubray, J.-M.; Garcia-Cazorla, À. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatr. Clin. N. Am. 2018, 65, 179–208. [Google Scholar] [CrossRef]
- Van Karnebeek, C.D.M.; Stockler, S. Treatable Inborn Errors of Metabolism Causing Intellectual Disability: A Systematic Literature Review. Mol. Genet. Metab. 2012, 105, 368–381. [Google Scholar] [CrossRef]
- Guthrie, R.; Susi, A. A Simple Phenylalanine Method for Detecting Phenyketonuria in Large Populations of Newborn Infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [CrossRef]
- Schulze, A.; Lindner, M.; Kohlmüller, D.; Olgemöller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization-Tandem Mass Spectrometry: Results, Outcome, and Implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.G.; Jungner, G.; World Health Organization. Principles and Practice of Screening for Disease; World Health Organization: Geneva, Switzerland, 1968. [Google Scholar]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Almannai, M.; Sutton, V.R. Newborn Screening: History, Current Status, and Future Directions. Pediatr. Clin. North Am. 2018, 65, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Vernon, H.J. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 2015, 169, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Mordaunt, D.; Cox, D.; Fuller, M. Metabolomics to Improve the Diagnostic Efficiency of Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 1195. [Google Scholar] [CrossRef] [Green Version]
- Jans, J.J.M.; Broeks, M.H.; Verhoeven-Duif, N.M. Metabolomics in Diagnostics of Inborn Metabolic Disorders. Curr. Opin. Syst. Biol. 2022, 29, 100409. [Google Scholar] [CrossRef]
- Cappuccio, G.; Pinelli, M.; Alagia, M.; Donti, T.; Day-Salvatore, D.-L.; Veggiotti, P.; Giorgis, V.D.; Lunghi, S.; Vari, M.S.; Striano, P.; et al. Biochemical Phenotyping Unravels Novel Metabolic Abnormalities and Potential Biomarkers Associated with Treatment of GLUT1 Deficiency with Ketogenic Diet. PLoS ONE 2017, 12, e0184022. [Google Scholar] [CrossRef] [Green Version]
- Ney, D.M.; Murali, S.G.; Stroup, B.M.; Nair, N.; Sawin, E.A.; Rohr, F.; Levy, H.L. Metabolomic Changes Demonstrate Reduced Bioavailability of Tyrosine and Altered Metabolism of Tryptophan via the Kynurenine Pathway with Ingestion of Medical Foods in Phenylketonuria. Mol. Genet. Metab. 2017, 121, 96–103. [Google Scholar] [CrossRef]
- Paine, R.S. The Variability in Manifestations of Untreated Patients with Phenylketonuria (Phenylpyruvic Aciduria). Pediatrics 1957, 20, 290–302. [Google Scholar] [CrossRef]
- De Groot, M.J.; Hoeksma, M.; Blau, N.; Reijngoud, D.J.; van Spronsen, F.J. Pathogenesis of Cognitive Dysfunction in Phenylketonuria: Review of Hypotheses. Mol. Genet. Metab. 2010, 99, S86–S89. [Google Scholar] [CrossRef]
- Burrage, L.C.; Thistlethwaite, L.; Stroup, B.M.; Sun, Q.; Miller, M.J.; Nagamani, S.C.; Craigen, W.; Scaglia, F.; Sutton, V.R.; Graham, B.; et al. Untargeted Metabolomic Profiling Reveals Multiple Pathway Perturbations and New Clinical Biomarkers in Urea Cycle Disorders. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1977–1986. [Google Scholar] [CrossRef]
- Mathis, T.; Poms, M.; Köfeler, H.; Gautschi, M.; Plecko, B.; Baumgartner, M.R.; Hochuli, M. Untargeted Plasma Metabolomics Identifies Broad Metabolic Perturbations in Glycogen Storage Disease Type I. J. Inherit. Metab. Dis. 2021, 45, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Sklirou, E.; Alodaib, A.N.; Dobrowolski, S.F.; Mohsen, A.-W.A.; Vockley, J. Physiological Perspectives on the Use of Triheptanoin as Anaplerotic Therapy for Long Chain Fatty Acid Oxidation Disorders. Front. Genet. 2020, 11, 598760. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.-J.C.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of Impaired Nitric Oxide Production in MELAS Syndrome with Citrulline and Arginine Supplementation. Mol. Genet. Metab. 2012, 105, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mew, N.A.; McCarter, R.; Daikhin, Y.; Lichter-Konecki, U.; Nissim, I.; Yudkoff, M.; Tuchman, M. Augmenting Ureagenesis in Patients with Partial Carbamyl Phosphate Synthetase 1 Deficiency with N-Carbamylglutamate. J. Pediatr. 2014, 165, 401–403.e3. [Google Scholar] [CrossRef] [Green Version]
- Heibel, S.K.; Mew, N.A.; Caldovic, L.; Daikhin, Y.; Yudkoff, M.; Tuchman, M. N-Carbamylglutamate Enhancement of Ureagenesis Leads to Discovery of a Novel Deleterious Mutation in a Newly Defined Enhancer of the NAGS Gene and to Effective Therapy. Hum. Mutat. 2011, 32, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Bandsma, R.H.J.; Rake, J.-P.; Visser, G.; Neese, R.A.; Hellerstein, M.K.; van Duyvenvoorde, W.; Princen, H.M.G.; Stellaard, F.; Smit, G.P.A.; Kuipers, F. Increased Lipogenesis and Resistance of Lipoproteins to Oxidative Modification in Two Patients with Glycogen Storage Disease Type 1a. J. Pediatr. 2002, 140, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Tebani, A.; Schmitz-Afonso, I.; Abily-Donval, L.; Héron, B.; Piraud, M.; Ausseil, J.; Brassier, A.; De Lonlay, P.; Zerimech, F.; Vaz, F.M.; et al. Urinary Metabolic Phenotyping of Mucopolysaccharidosis Type I Combining Untargeted and Targeted Strategies with Data Modeling. Clin. Chim. Acta 2017, 475, 7–14. [Google Scholar] [CrossRef]
- Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Héron, B.; Piraud, M.; Ausseil, J.; Zerimech, F.; Gonzalez, B.; Marret, S.; Afonso, C.; et al. Unveiling Metabolic Remodeling in Mucopolysaccharidosis Type III through Integrative Metabolomics and Pathway Analysis. J. Transl. Med. 2018, 16, 248. [Google Scholar] [CrossRef]
- Tebani, A.; Abily-Donval, L.; Schmitz-Afonso, I.; Piraud, M.; Ausseil, J.; Zerimech, F.; Pilon, C.; Pereira, T.; Marret, S.; Afonso, C.; et al. Analysis of Mucopolysaccharidosis Type VI through Integrative Functional Metabolomics. Int. J. Mol. Sci. 2019, 20, 446. [Google Scholar] [CrossRef] [Green Version]
- Reed, L.K.; Baer, C.F.; Edison, A.S. Considerations When Choosing a Genetic Model Organism for Metabolomics Studies. Curr. Opin. Chem. Biol. 2017, 36, 7–14. [Google Scholar] [CrossRef]
- Fu, H.; Meadows, A.S.; Ware, T.; Mohney, R.P.; McCarty, D.M. Near-Complete Correction of Profound Metabolomic Impairments Corresponding to Functional Benefit in MPS IIIB Mice after IV RAAV9-HNAGLU Gene Delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 792–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.-H.; Xia, Z.-X.; Guo, J.-L.; Xiao, L.-L.; Zhang, Y.-J. Metabolomics Analysis Reveals Perturbations of Cerebrocortical Metabolic Pathways in the Pahenu2 Mouse Model of Phenylketonuria. CNS Neurosci. Ther. 2020, 26, 486–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Valencia, I.; Good, L.B.; Ma, Q.; Duarte, J.; Bottiglieri, T.; Sinton, C.M.; Heilig, C.W.; Pascual, J.M. Glut1 Deficiency (G1D): Epilepsy and Metabolic Dysfunction in a Mouse Model of the Most Common Human Phenotype. Neurobiol. Dis. 2012, 48, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Hijmans, B.S.; Boss, A.; van Dijk, T.H.; Soty, M.; Wolters, H.; Mutel, E.; Groen, A.K.; Derks, T.G.J.; Mithieux, G.; Heerschap, A.; et al. Hepatocytes Contribute to Residual Glucose Production in a Mouse Model for Glycogen Storage Disease Type Ia. Hepatol. Baltim. Md 2017, 66, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Norman, B.P.; Davison, A.S.; Hughes, J.H.; Sutherland, H.; Wilson, P.J.M.; Berry, N.G.; Hughes, A.T.; Milan, A.M.; Jarvis, J.C.; Roberts, N.B.; et al. Metabolomic Studies in the Inborn Error of Metabolism Alkaptonuria Reveal New Biotransformations in Tyrosine Metabolism. Genes Dis. 2021, 9, 3. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Vaz, F.M.; Ferdinandusse, S.; van Kuilenburg, A.B.P.; Kemp, S.; van Karnebeek, C.D.; Waterham, H.R.; Houtkooper, R.H. Translational Metabolism: A Multidisciplinary Approach towards Precision Diagnosis of Inborn Errors of Metabolism in the Omics Era. J. Inherit. Metab. Dis. 2019, 42, 197–208. [Google Scholar] [CrossRef]
- Falk, M.J.; Zhang, Z.; Rosenjack, J.R.; Nissim, I.; Daikhin, D.; Nissim, I.; Sedensky, M.M.; Yudkoff, M.; Morgan, P.G. Metabolic Pathway Profiling of Mitochondrial Respiratory Chain Mutants in C. Elegans. Mol. Genet. Metab. 2008, 93, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Vergano, S.S.; Rao, M.; McCormack, S.; Ostrovsky, J.; Clarke, C.; Preston, J.; Bennett, M.J.; Yudkoff, M.; Xiao, R.; Falk, M.J. In Vivo Metabolic Flux Profiling with Stable Isotopes Discriminates Sites and Quantifies Effects of Mitochondrial Dysfunction in C. Elegans. Mol. Genet. Metab. 2014, 111, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.A.; Ostrovsky, J.; Rao, M.; Dingley, S.D.; Polyak, E.; Yudkoff, M.; Xiao, R.; Bennett, M.J.; Falk, M.J. Propionyl-CoA Carboxylase Pcca-1 and Pccb-1 Gene Deletions in Caenorhabditis Elegans Globally Impair Mitochondrial Energy Metabolism. J. Inherit. Metab. Dis. 2018, 41, 157–168. [Google Scholar] [CrossRef]
- Salzer, L.; Witting, M. Quo Vadis Caenorhabditis Elegans Metabolomics-A Review of Current Methods and Applications to Explore Metabolism in the Nematode. Metabolites 2021, 11, 284. [Google Scholar] [CrossRef]
- Čuperlović-Culf, M.; Barnett, D.A.; Culf, A.S.; Chute, I. Cell Culture Metabolomics: Applications and Future Directions. Drug Discov. Today 2010, 15, 610–621. [Google Scholar] [CrossRef]
- Van Rijn, J.M.; Ardy, R.C.; Kuloğlu, Z.; Härter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.J.; van Hoesel, M.; Kansu, A.; van Vugt, A.H.M.; Thian, M.; et al. Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency. Gastroenterology 2018, 155, 130–143.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammage, P.A.; Van Haute, L.; Minczuk, M. Engineered MtZFNs for Manipulation of Human Mitochondrial DNA Heteroplasmy. In Mitochondrial DNA: Methods and Protocols; McKenzie, M., Ed.; Springer: New York, NY, USA, 2016; pp. 145–162. ISBN 978-1-4939-3040-1. [Google Scholar]
- Bao, X.R.; Ong, S.-E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial Dysfunction Remodels One-Carbon Metabolism in Human Cells. eLife 2016, 5, e10575. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Schmidt, C.; Gammage, P.A.; Dugourd, A.; Blacker, T.; Chew, S.P.; Saez-Rodriguez, J.; O’Neill, J.S.; Szabadkai, G.; Minczuk, M.; et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction. Mol. Cell 2018, 69, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, P.Y.; Harrison, P.T.; Davidson, A.J.; Hollywood, J.A. In Vitro and In Vivo Models to Study Nephropathic Cystinosis. Cells 2021, 11, 6. [Google Scholar] [CrossRef]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.M.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, Encoding the Mitochondrial Citrate Carrier, Causes Combined D-2- and L-2-Hydroxyglutaric Aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Pop, A.; Williams, M.; Struys, E.A.; Monné, M.; Jansen, E.E.W.; De Grassi, A.; Kanhai, W.A.; Scarcia, P.; Ojeda, M.R.F.; Porcelli, V.; et al. An Overview of Combined D-2- and L-2-Hydroxyglutaric Aciduria: Functional Analysis of CIC Variants. J. Inherit. Metab. Dis. 2018, 41, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, L.; Theimer, J.; Wingert, V.; Klotz, K.; Bierschenk, I.; Nitschke, R.; Spiekerkoetter, U.; Grünert, S.C. Metabolic Profiling in Human Fibroblasts Enables Subtype Clustering in Glycogen Storage Disease. Front. Endocrinol. 2020, 11, 579981. [Google Scholar] [CrossRef]
- Ni, M.; Solmonson, A.; Pan, C.; Yang, C.; Li, D.; Notzon, A.; Cai, L.; Guevara, G.; Zacharias, L.G.; Faubert, B.; et al. Functional Assessment of Lipoyltransferase-1 Deficiency in Cells, Mice, and Humans. Cell Rep. 2019, 27, 1376–1386.e6. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Black, L.F.; Pan, C.; Vu, H.; Pei, J.; Ko, B.; Cai, L.; Solmonson, A.; Yang, C.; Nugent, K.M.; et al. Metabolic Impact of Pathogenic Variants in the Mitochondrial Glutamyl-TRNA Synthetase EARS2. J. Inherit. Metab. Dis. 2021, 44, 949–960. [Google Scholar] [CrossRef]
- Radenkovic, S.; Bird, M.J.; Emmerzaal, T.L.; Wong, S.Y.; Felgueira, C.; Stiers, K.M.; Sabbagh, L.; Himmelreich, N.; Poschet, G.; Windmolders, P.; et al. The Metabolic Map into the Pathomechanism and Treatment of PGM1-CDG. Am. J. Hum. Genet. 2019, 104, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, M.; Scott, D.A.; Losfeld, M.-E.; Freeze, H.H. The Metabolic Origins of Mannose in Glycoproteins. J. Biol. Chem. 2014, 289, 6751–6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morava, E.; Schatz, U.A.; Torring, P.M.; Abbott, M.-A.; Baumann, M.; Brasch-Andersen, C.; Chevalier, N.; Dunkhase-Heinl, U.; Fleger, M.; Haack, T.B.; et al. Impaired Glucose-1,6-Biphosphate Production Due to Bi-Allelic PGM2L1 Mutations Is Associated with a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2021, 108, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, S.; Martinelli, D.; Zhang, Y.; Preston, G.J.; Maiorana, A.; Terracciano, A.; Dentici, M.L.; Pisaneschi, E.; Novelli, A.; Ranatunga, W.; et al. TRAPPC9-CDG: A Novel Congenital Disorder of Glycosylation with Dysmorphic Features and Intellectual Disability. Genet. Med. 2021, 24, 894–904. [Google Scholar] [CrossRef]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Driesen, K.; Witters, P. Understanding Inborn Errors of Metabolism through Metabolomics. Metabolites 2022, 12, 398. https://doi.org/10.3390/metabo12050398
Driesen K, Witters P. Understanding Inborn Errors of Metabolism through Metabolomics. Metabolites. 2022; 12(5):398. https://doi.org/10.3390/metabo12050398
Chicago/Turabian StyleDriesen, Karen, and Peter Witters. 2022. "Understanding Inborn Errors of Metabolism through Metabolomics" Metabolites 12, no. 5: 398. https://doi.org/10.3390/metabo12050398