
Normalizing and Correcting Variable and Complex LC–MS Metabolomic Data with the R Package pseudoDrift


Abstract
1. Introduction
2. Results and Discussion
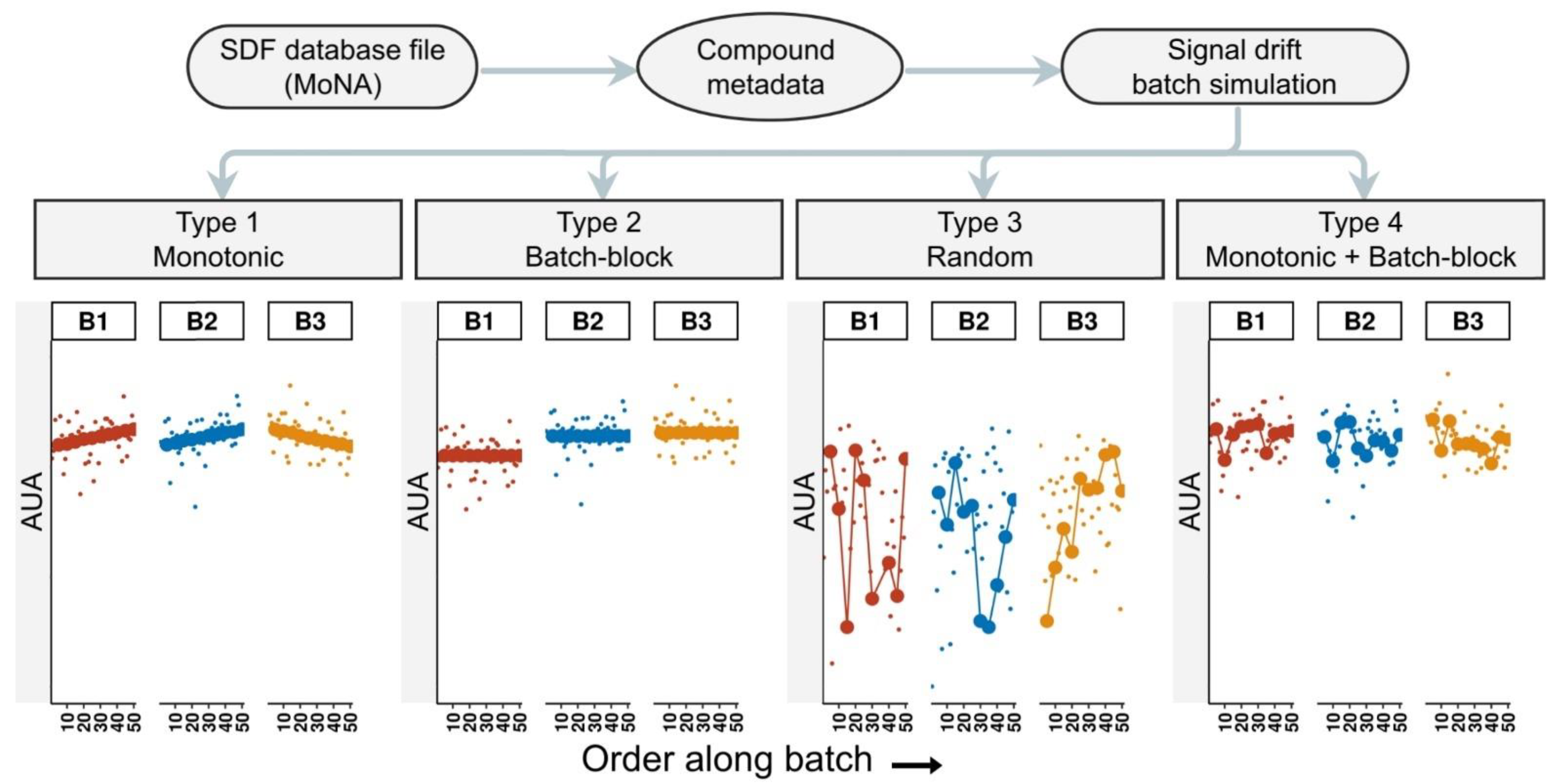
2.1. Simulating Data with pseudoDrift
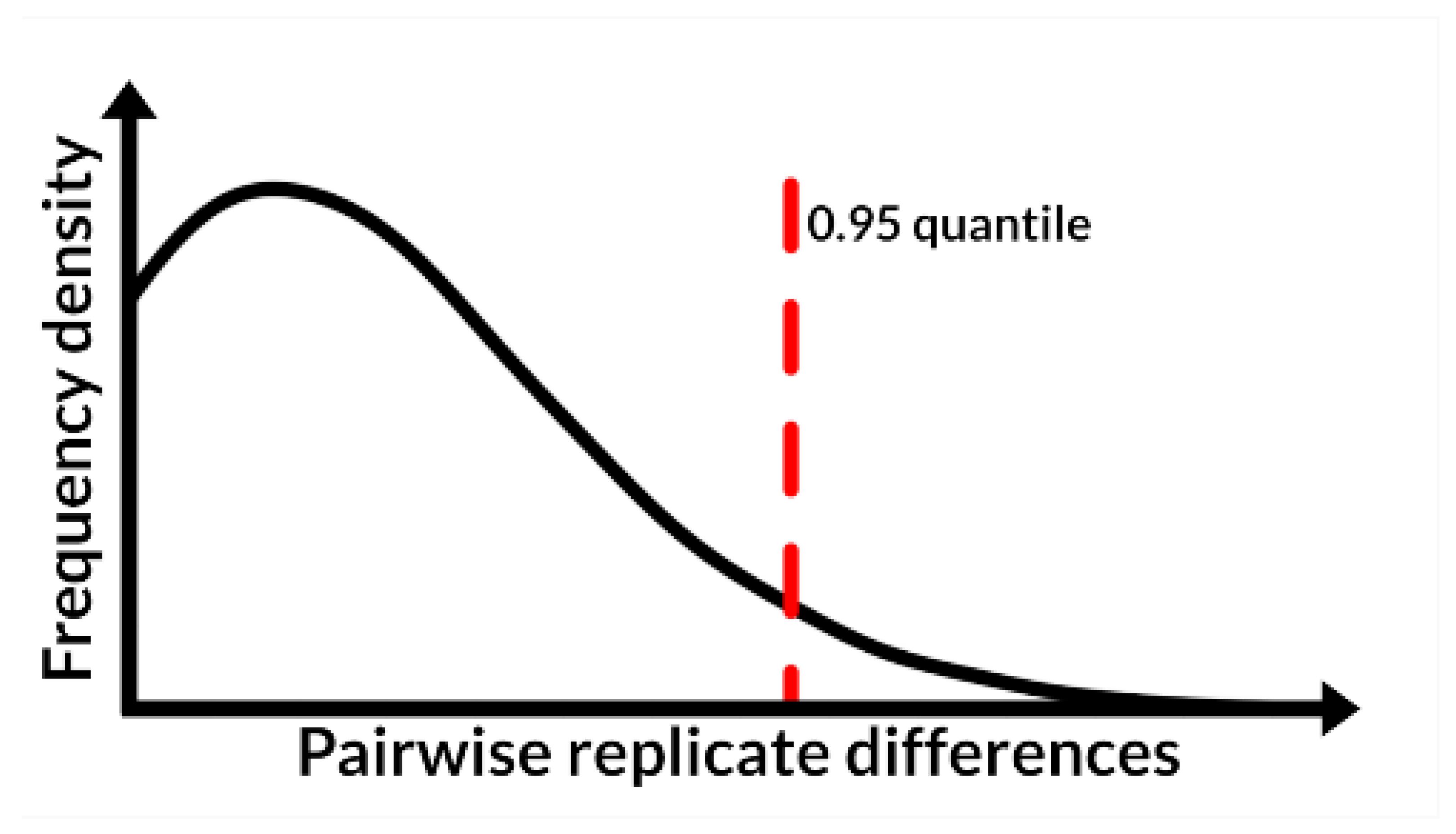
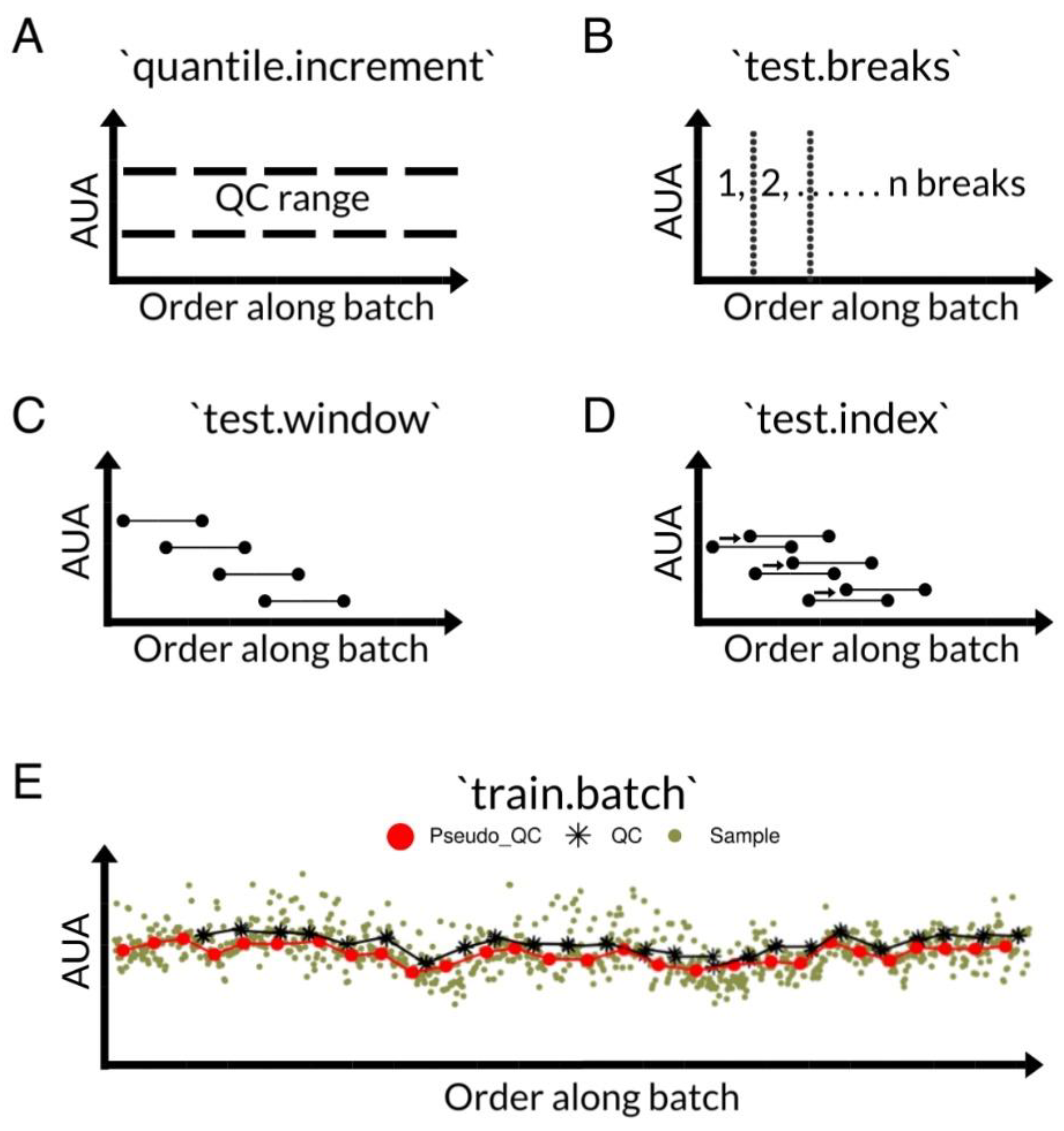
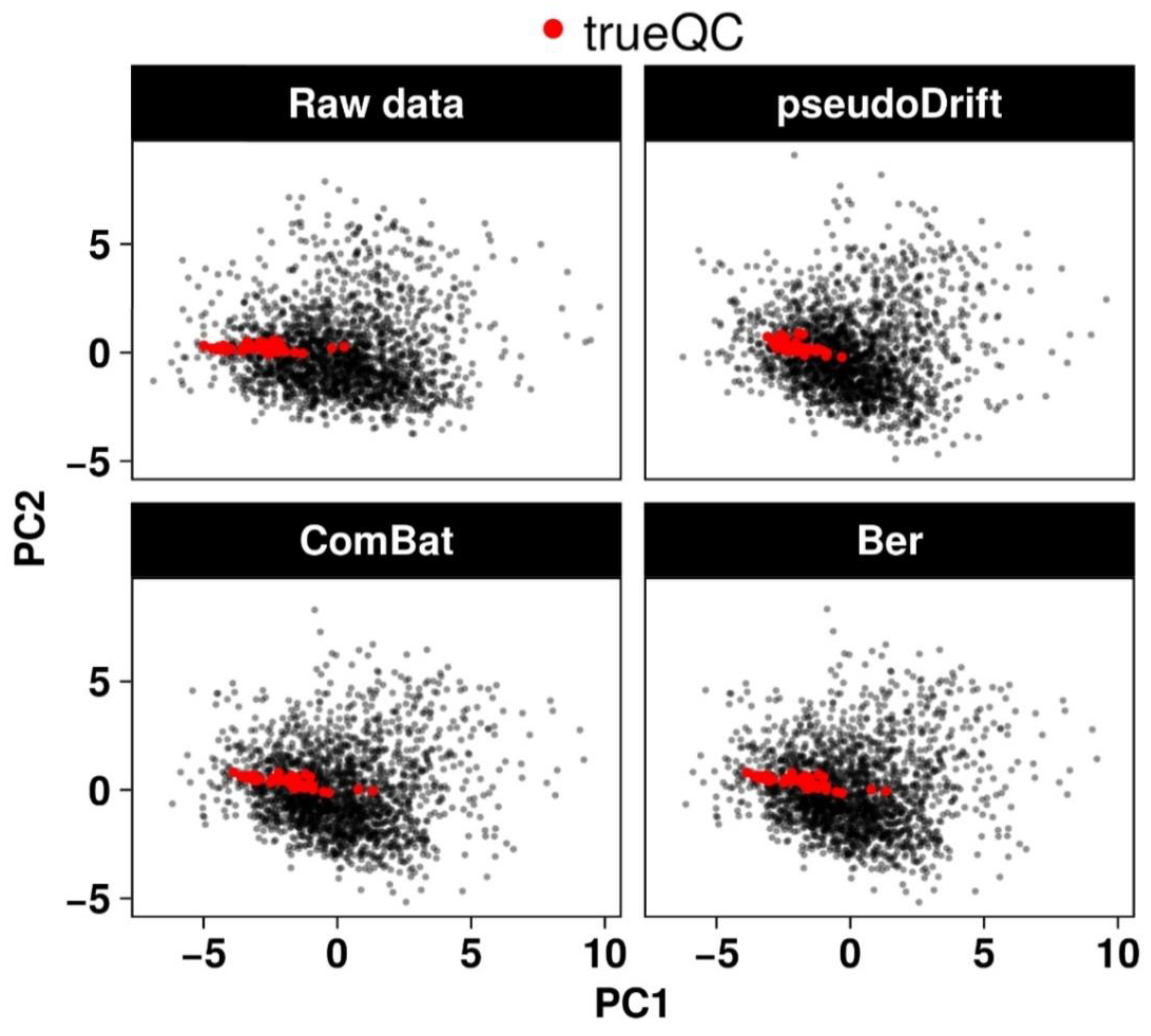
2.2. Performance Evaluation of the pseudoDrift Analysis Workflow
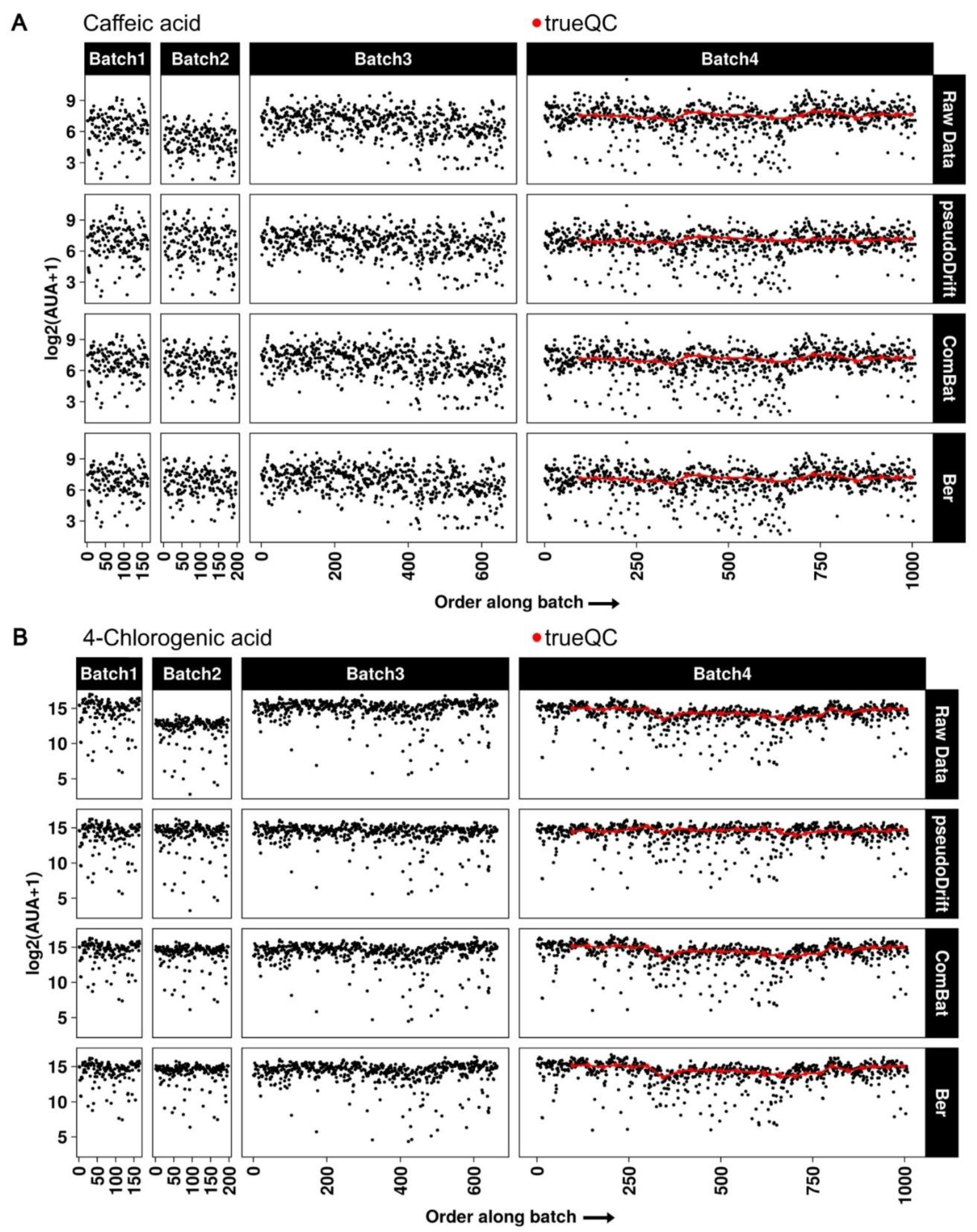
2.3. Maize LC–MS Phenolic Data Analysis with pseudoDrift
3. Materials and Methods
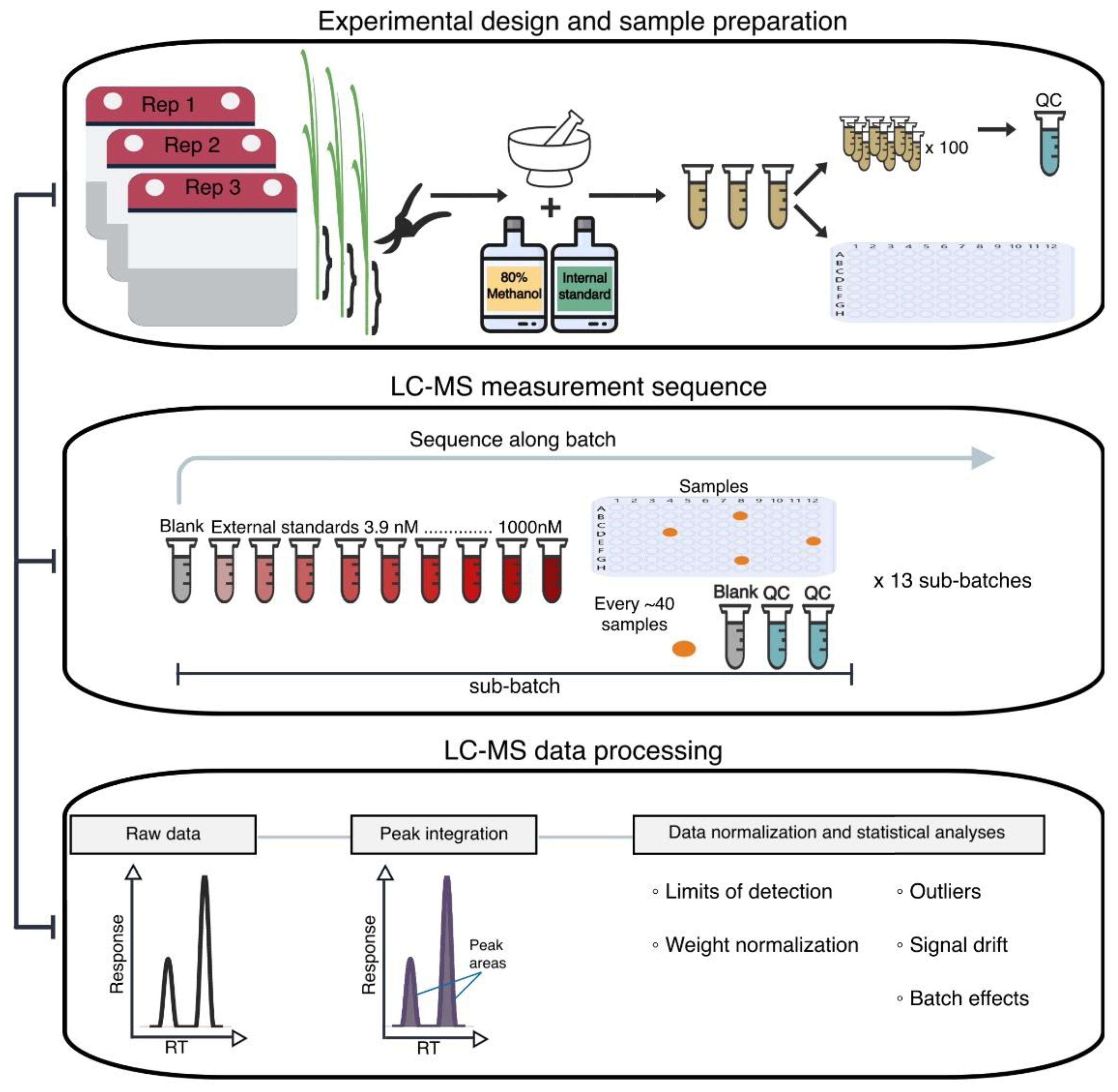
3.1. Plant Material and Experimental Design
3.2. Reagents for Stock and Working Solutions
3.3. Preparation of Stem Tissue Extracts and QC Samples
3.4. LC–MS Data Acquisition
3.5. PseudoDrift Workflow
3.6. LC–MS Data Normalization and Processing with pseudoDrift
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted Metabolomics. Curr. Protoc. Mol. Biol. 2012, 98, 30.2.1–30.2.24. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Saand, M.A.; Huang, L.; Abdelaal, W.B.; Zhang, J.; Wu, Y.; Li, J.; Sirohi, M.H.; Wang, F. Applications of Multi-Omics Technologies for Crop Improvement. Front. Plant Sci. 2021, 12, 1846. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, Transcriptome and Proteome: The Rise of Omics Data and Their Integration in Biomedical Sciences. Brief. Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for Plant Improvement: Status and Prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef]
- Ranum, P.; Peña-Rosas, J.P.; Garcia-Casal, M.N. Global Maize Production, Utilization, and Consumption. Ann. N. Y. Acad. Sci. 2014, 1312, 105–112. [Google Scholar] [CrossRef]
- Medeiros, D.B.; Brotman, Y.; Fernie, A.R. The Utility of Metabolomics as a Tool to Inform Maize Biology. Plant Commun. 2021, 2, 100187. [Google Scholar] [CrossRef]
- Sánchez-Illana, Á.; Piñeiro-Ramos, J.D.; Sanjuan-Herráez, J.D.; Vento, M.; Quintás, G.; Kuligowski, J. Evaluation of Batch Effect Elimination Using Quality Control Replicates in LC-MS Metabolite Profiling. Anal. Chim. Acta 2018, 1019, 38–48. [Google Scholar] [CrossRef]
- Wehrens, R.; Hageman, J.A.; van Eeuwijk, F.; Kooke, R.; Flood, P.J.; Wijnker, E.; Keurentjes, J.J.B.; Lommen, A.; van Eekelen, H.D.L.M.; Hall, R.D.; et al. Improved Batch Correction in Untargeted MS-Based Metabolomics. Metabolomics 2016, 12, 88. [Google Scholar] [CrossRef]
- Kuligowski, J.; Sánchez-Illana, Á.; Sanjuán-Herráez, D.; Vento, M.; Quintás, G. Intra-Batch Effect Correction in Liquid Chromatography-Mass Spectrometry Using Quality Control Samples and Support Vector Regression (QC-SVRC). Analyst 2015, 140, 7810–7817. [Google Scholar] [CrossRef]
- Han, W.; Li, L. Evaluating and Minimizing Batch Effects in Metabolomics. Mass Spectrom. Rev. 2020, 1–22. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and Considerations for the Use of System Suitability and Quality Control Samples in Mass Spectrometry Assays Applied in Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.A.; Broadhurst, D.I.; Davidson, R.L.; Viant, M.R. Characterising and Correcting Batch Variation in an Automated Direct Infusion Mass Spectrometry (DIMS) Metabolomics Workflow. Anal. Bioanal. Chem. 2013, 405, 5147–5157. [Google Scholar] [CrossRef] [PubMed]
- Rusilowicz, M.; Dickinson, M.; Charlton, A.; O’Keefe, S.; Wilson, J. A Batch Correction Method for Liquid Chromatography–Mass Spectrometry Data That Does Not Depend on Quality Control Samples. Metabolomics 2016, 12, 56. [Google Scholar] [CrossRef]
- Bararpour, N.; Gilardi, F.; Carmeli, C.; Sidibe, J.; Ivanisevic, J.; Caputo, T.; Augsburger, M.; Grabherr, S.; Desvergne, B.; Guex, N.; et al. DBnorm as an R Package for the Comparison and Selection of Appropriate Statistical Methods for Batch Effect Correction in Metabolomic Studies. Sci. Rep. 2021, 11, 5657. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Trieglaff, O.; Pfeifer, N.; Gröpl, C.; Kohlbacher, O.; Reinert, K. LC-MSsim – a Simulation Software for Liquid Chromatography Mass Spectrometry Data. BMC Bioinformatics 2008, 9, 423. [Google Scholar] [CrossRef] [PubMed]
- Kösters, M.; Leufken, J.; Leidel, S.A. SMITER-A Python Library for the Simulation of LC-MS/MS Experiments. Genes 2021, 12, 396. [Google Scholar] [CrossRef]
- Bielow, C.; Aiche, S.; Andreotti, S.; Reinert, K. MSSimulator: Simulation of Mass Spectrometry Data. J. Proteome Res. 2011, 10, 2922–2929. [Google Scholar] [CrossRef][Green Version]
- Noyce, A.B.; Smith, R.; Dalgleish, J.; Taylor, R.M.; Erb, K.C.; Okuda, N.; Prince, J.T. Mspire-Simulator: LC-MS Shotgun Proteomic Simulator for Creating Realistic Gold Standard Data. J. Proteome Res. 2013, 12, 5742–5749. [Google Scholar] [CrossRef]
- MassBank of North America. Available online: https://mona.fiehnlab.ucdavis.edu/ (accessed on 8 January 2022).
- Hansey, C.N.; Johnson, J.M.; Sekhon, R.S.; Kaeppler, S.M.; de Leon, N. Genetic Diversity of a Maize Association Population with Restricted Phenology. Crop Sci. 2011, 51, 704–715. [Google Scholar] [CrossRef]
- Mazaheri, M.; Heckwolf, M.; Vaillancourt, B.; Gage, J.L.; Burdo, B.; Heckwolf, S.; Barry, K.; Lipzen, A.; Ribeiro, C.B.; Kono, T.J.Y.; et al. Genome-Wide Association Analysis of Stalk Biomass and Anatomical Traits in Maize. BMC Plant Biol. 2019, 19, 45. [Google Scholar] [CrossRef]
- Grubbs, F.E. Sample Criteria for Testing Outlying Observations. Ann. Math. Stat. 1950, 21, 27–58. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinforma. Oxf. Engl. 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Giordan, M. A Two-Stage Procedure for the Removal of Batch Effects in Microarray Studies. Stat. Biosci. 2014, 6, 73–84. [Google Scholar] [CrossRef]
- Cocuron, J.C.; Casas, M.I.; Yang, F.; Grotewold, E.; Alonso, A.P. Beyond the Wall: High-Throughput Quantification of Plant Soluble and Cell-Wall Bound Phenolics by Liquid Chromatography Tandem Mass Spectrometry. J. Chromatogr. A 2019, 1589, 93–104. [Google Scholar] [CrossRef]
- Jankevics, A.; Lloyd, G.R.; Weber, R.J.M. Pmp: Peak Matrix Processing and Signal Batch Correction for Metabolomics Datasets. Available online: https://bioconductor.org/packages/pmp/ (accessed on 6 January 2022).
- Cao, Y.E.; Horan, K.; Backman, T.; Girke, T. ChemmineR: Cheminformatics Toolkit for R. Available online: https://bioconductor.org/packages/ChemmineR/ (accessed on 24 March 2022).
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 24 March 2022).
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “Ggplot2”. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 24 March 2022).
- Dowle, M.; Srinivasan, A.; Gorecki, J.; Chirico, M.; Stetsenko, P.; Short, T.; Lianoglou, S.; Antonyan, E.; Bonsch, M.; Parsonage, H.; et al. Data. Table: Extension of “Data.Frame”. Available online: https://CRAN.R-project.org/package=data.table (accessed on 24 March 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; R Core Team; et al. Caret: Classification and Regression Training. Available online: https://CRAN.R-project.org/package=caret (accessed on 24 March 2022).
- Dong, N.-Q.; Lin, H.-X. Contribution of Phenylpropanoid Metabolism to Plant Development and Plant–Environment Interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Parihar, A.; Grotewold, E.; Doseff, A.I. Flavonoid Dietetics: Mechanisms and Emerging Roles of Plant Nutraceuticals. In Pigments in Fruits and Vegetables: Genomics and Dietetics; Chen, C., Ed.; Springer: New York, NY, USA, 2015; pp. 93–126. ISBN 978-1-4939-2356-4. [Google Scholar]
- Jiang, N.; Doseff, A.I.; Grotewold, E. Flavones: From Biosynthesis to Health Benefits. Plants Basel Switz. 2016, 5, 27. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Batch | Num. Samples | External Standard | Internal Standard | QC Samples Represented |
|---|---|---|---|---|
| B1 | 165 | Yes | No | No |
| B2 | 198 | Yes | No | No |
| B3 | 663 | Yes | No | No |
| B4 | 1008 | Yes | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, J.; Gomez-Cano, L.; Grotewold, E.; de Leon, N. Normalizing and Correcting Variable and Complex LC–MS Metabolomic Data with the R Package pseudoDrift. Metabolites 2022, 12, 435. https://doi.org/10.3390/metabo12050435
Rodriguez J, Gomez-Cano L, Grotewold E, de Leon N. Normalizing and Correcting Variable and Complex LC–MS Metabolomic Data with the R Package pseudoDrift. Metabolites. 2022; 12(5):435. https://doi.org/10.3390/metabo12050435
Chicago/Turabian StyleRodriguez, Jonas, Lina Gomez-Cano, Erich Grotewold, and Natalia de Leon. 2022. "Normalizing and Correcting Variable and Complex LC–MS Metabolomic Data with the R Package pseudoDrift" Metabolites 12, no. 5: 435. https://doi.org/10.3390/metabo12050435
APA StyleRodriguez, J., Gomez-Cano, L., Grotewold, E., & de Leon, N. (2022). Normalizing and Correcting Variable and Complex LC–MS Metabolomic Data with the R Package pseudoDrift. Metabolites, 12(5), 435. https://doi.org/10.3390/metabo12050435